13535071
Beschreibung
Mindmap von Maria Fernanda Bravo Sanchez, aktualisiert more than 1 year ago
|
|
Erstellt von Maria Fernanda Bravo Sanchez
vor mehr als 6 Jahre
|
|
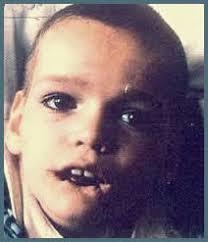
6. Retraso mental
- Sindromde de Down
- El síndrome de Down es la más común y fácil de reconocer
de todas las condiciones asociadas con la discapacidad
intelectual. Esta condición (antes conocida como retraso
mental) es el resultado de una anormalidad de los
cromosomas: por alguna razón inexplicable una desviación
en el desarrollo de las células resulta en la producción de
47 cromosomas en lugar de las 46 que se consideran
normales. El cromosoma adicional cambia totalmente el
desarrollo ordenado del cuerpo y cerebro. En la mayor
parte de los casos, el diagnóstico del síndrome de Down se
hace de acuerdo a los resultados de una prueba de
cromosomas que es suministrada poco después del
nacimiento del niño.
- Hay más de 50 síntomas reconocidos del síndrome de Down,
pero es raro encontrar una persona con todos o una gran
cantidad de éstos. Cada bebé con síndrome de Down es
diferente. Algunos nacen con pocos de los rasgos listados
abajo y otros con más. Algunas de estas características pueden
incluir:
- Falta de tono muscular; • Ojos alargados, con el cutis
pliegado en el rabillo del ojo; • Hiperflexibilidad (la
habilidad de extender excesivamente las coyunturas); •
Manos chicas y anchas con una sola arruga en la palma
de una o ambas manos; • Pies anchos con los dedos
cortos; • El puente de la nariz plano; • Orejas pequeñas,
en la parte inferior de la cabeza; • Cuello corto y cabeza
pequeña.
- Hay dos tipos de pruebas para el síndrome de Down
que se pueden realizar antes de que nazca un bebé:
pruebas de detección y pruebas diagnósticas. Las
pruebas de detección prenatales calculan la
posibilidad de que el feto tenga el síndrome de Down.
Las pruebas diagnósticas pueden proporcionar un
diagnóstico definitivo con casi el 100% de precisión.
- Hay dos tipos de pruebas para el síndrome de Down
que se pueden realizar antes de que nazca un bebé:
pruebas de detección y pruebas diagnósticas. Las
pruebas de detección prenatales calculan la
posibilidad de que el feto tenga el síndrome de Down.
Las pruebas diagnósticas pueden proporcionar un
diagnóstico definitivo con casi el 100% de precisión.
- Falta de tono muscular; • Ojos alargados, con el cutis
pliegado en el rabillo del ojo; • Hiperflexibilidad (la
habilidad de extender excesivamente las coyunturas); •
Manos chicas y anchas con una sola arruga en la palma
de una o ambas manos; • Pies anchos con los dedos
cortos; • El puente de la nariz plano; • Orejas pequeñas,
en la parte inferior de la cabeza; • Cuello corto y cabeza
pequeña.
- Hay más de 50 síntomas reconocidos del síndrome de Down,
pero es raro encontrar una persona con todos o una gran
cantidad de éstos. Cada bebé con síndrome de Down es
diferente. Algunos nacen con pocos de los rasgos listados
abajo y otros con más. Algunas de estas características pueden
incluir:
- El síndrome de Down es la más común y fácil de reconocer
de todas las condiciones asociadas con la discapacidad
intelectual. Esta condición (antes conocida como retraso
mental) es el resultado de una anormalidad de los
cromosomas: por alguna razón inexplicable una desviación
en el desarrollo de las células resulta en la producción de
47 cromosomas en lugar de las 46 que se consideran
normales. El cromosoma adicional cambia totalmente el
desarrollo ordenado del cuerpo y cerebro. En la mayor
parte de los casos, el diagnóstico del síndrome de Down se
hace de acuerdo a los resultados de una prueba de
cromosomas que es suministrada poco después del
nacimiento del niño.
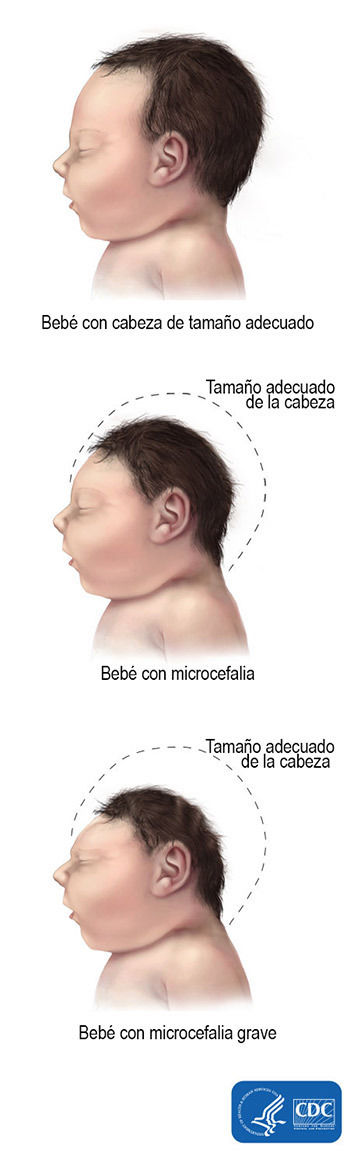
- Microcefalia
- La microcefalia es una afección en la cual la cabeza del bebé es mucho más
pequeña de lo esperado. Durante el embarazo, la cabeza del bebé aumenta
de tamaño porque el cerebro del bebé crece. La microcefalia puede ocurrir
porque el cerebro del bebé no se ha desarrollado adecuadamente durante el
embarazo o dejó de crecer después del nacimiento, lo que produce un
tamaño de la cabeza más pequeño. La microcefalia puede ser una afección
aislada, lo que significa que puede ocurrir sin que ocurran otros defectos
graves, o puede presentarse en combinación con otros defectos de
nacimiento graves.
- La microcefalia grave es una forma más grave y extrema de esta
afección, en la cual la cabeza del bebé es mucho más pequeña de
lo esperado. La microcefalia grave puede ocurrir cuando el cerebro
del bebé no se desarrolla adecuadamente durante el embarazo, o
cuando el cerebro se empieza a desarrollar correctamente, pero
en algún momento del embarazo ocurren daños y el cerebro deja
de crecer.
- Los bebés con microcefalia pueden tener una gama de problemas
adicionales, dependiendo de lo grave que sea esa afección. La
microcefalia se ha asociado a los siguientes problemas: Convulsiones.
Retraso en el desarrollo, como problemas del habla y con otros
indicadores del desarrollo (como sentarse, pararse y caminar).
Discapacidad intelectual (disminución de la capacidad para aprender y
funcionar en la vida diaria). Problemas con el movimiento y el equilibrio.
Problemas para alimentarse, como dificultad para tragar. Pérdida de la
audición.
- Causas y factores de riesgo Se desconocen las causas de la
microcefalia en la mayoría de los bebés. Algunos bebés tienen
microcefalia por cambios en sus genes. Otras causas de la
microcefalia, incluso de la microcefalia grave, pueden incluir las
siguientes exposiciones durante el embarazo: Ciertas infecciones
como la rubéola, la toxoplasmosis o por el citomegalovirus. La
desnutrición grave, es decir la falta de nutrientes o no alimentarse
lo suficiente. La exposición a sustancias dañinas, como alcohol,
ciertos medicamentos o sustancias químicas tóxicas. La interrupción
del flujo de sangre al cerebro del bebé durante su desarrollo. Los
investigadores están estudiando un posible vínculo entre la
infección por el virus del Zika y la microcefalia. Los CDC continúan
estudiando los defectos congénitos, como la microcefalia, y la forma
de prevenirlos. Si usted está embarazada o planea quedar
embarazada, pregúntele a su médico cómo puede aumentar su
probabilidad de tener un bebé saludable.
- Causas y factores de riesgo Se desconocen las causas de la
microcefalia en la mayoría de los bebés. Algunos bebés tienen
microcefalia por cambios en sus genes. Otras causas de la
microcefalia, incluso de la microcefalia grave, pueden incluir las
siguientes exposiciones durante el embarazo: Ciertas infecciones
como la rubéola, la toxoplasmosis o por el citomegalovirus. La
desnutrición grave, es decir la falta de nutrientes o no alimentarse
lo suficiente. La exposición a sustancias dañinas, como alcohol,
ciertos medicamentos o sustancias químicas tóxicas. La interrupción
del flujo de sangre al cerebro del bebé durante su desarrollo. Los
investigadores están estudiando un posible vínculo entre la
infección por el virus del Zika y la microcefalia. Los CDC continúan
estudiando los defectos congénitos, como la microcefalia, y la forma
de prevenirlos. Si usted está embarazada o planea quedar
embarazada, pregúntele a su médico cómo puede aumentar su
probabilidad de tener un bebé saludable.
- Los bebés con microcefalia pueden tener una gama de problemas
adicionales, dependiendo de lo grave que sea esa afección. La
microcefalia se ha asociado a los siguientes problemas: Convulsiones.
Retraso en el desarrollo, como problemas del habla y con otros
indicadores del desarrollo (como sentarse, pararse y caminar).
Discapacidad intelectual (disminución de la capacidad para aprender y
funcionar en la vida diaria). Problemas con el movimiento y el equilibrio.
Problemas para alimentarse, como dificultad para tragar. Pérdida de la
audición.
- La microcefalia grave es una forma más grave y extrema de esta
afección, en la cual la cabeza del bebé es mucho más pequeña de
lo esperado. La microcefalia grave puede ocurrir cuando el cerebro
del bebé no se desarrolla adecuadamente durante el embarazo, o
cuando el cerebro se empieza a desarrollar correctamente, pero
en algún momento del embarazo ocurren daños y el cerebro deja
de crecer.
- La microcefalia es una afección en la cual la cabeza del bebé es mucho más
pequeña de lo esperado. Durante el embarazo, la cabeza del bebé aumenta
de tamaño porque el cerebro del bebé crece. La microcefalia puede ocurrir
porque el cerebro del bebé no se ha desarrollado adecuadamente durante el
embarazo o dejó de crecer después del nacimiento, lo que produce un
tamaño de la cabeza más pequeño. La microcefalia puede ser una afección
aislada, lo que significa que puede ocurrir sin que ocurran otros defectos
graves, o puede presentarse en combinación con otros defectos de
nacimiento graves.
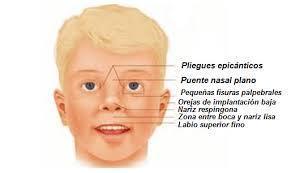
- Lesh-Nyhan
- El sindrome de Lesch-Nyhan es un trastorno que se transmite de
padres a hijos (hereditario). Afecta la forma como el cuerpo
produce y descompone las purinas. Las purina son una parte
normal del tejido humano y ayudan a conformar la constitución
genética del cuerpo. También se encuentran en muchos alimentos
diferentes.
- El síndrome de Lesch-Nyhan se transmite como un rasgo ligado al sexo, o
un rasgo ligado al cromosoma X, y en su mayoría se presenta en niños
varones. Las personas con este síndrome no tienen o tienen muy poca
cantidad de una enzima llamada hipoxantina guanina
fosforribosiltransferasa (HPRT, por sus siglas en inglés) una sustancia que
el cuerpo necesita para reciclar las purinas. Sin esta, se acumulan en el
cuerpo niveles anormalmente altos de ácido úrico.
- Demasiado ácido úrico puede provocar una hinchazón seudogotosa en
algunas de las articulaciones. En algunos casos, se desarrollan cálculos
renales y vesicales. Las personas con Lesch-Nyhan tienen retraso del
desarrollo motor seguido de movimientos anormales y aumento de los
reflejos. Un rasgo llamativo del síndrome de Lesch-Nyhan es el
comportamiento autodestructivo, lo que incluye morderse las yemas de
los dedos y los labios. Se desconoce la forma como la enfermedad causa
estos problemas.
- Puede haber antecedentes familiares de esta afección. El proveedor de
atención médica llevará a cabo un examen físico. El examen puede
mostrar: Reflejos demasiado exagerados Espasticidad (tener espasmos)
Los exámenes de sangre y orina pueden mostrar niveles altos de ácido
úrico. Una biopsia de piel puede mostrar disminución de los niveles de la
enzima HGP.
- Tratamiento No existe un tratamiento específico para el síndrome
de Lesch-Nyhan. El medicamento para tratar la gota puede bajar los
niveles de ácido úrico. Sin embargo, el tratamiento no mejora el
pronóstico neurológico (por ejemplo, tener aumento de los reflejos y
los espasmos). Algunos síntomas se pueden aliviar con estos
medicamentos: Carbidopa/levodopa Diazepan Fenobarbital
Haloperidol
- Tratamiento No existe un tratamiento específico para el síndrome
de Lesch-Nyhan. El medicamento para tratar la gota puede bajar los
niveles de ácido úrico. Sin embargo, el tratamiento no mejora el
pronóstico neurológico (por ejemplo, tener aumento de los reflejos y
los espasmos). Algunos síntomas se pueden aliviar con estos
medicamentos: Carbidopa/levodopa Diazepan Fenobarbital
Haloperidol
- Puede haber antecedentes familiares de esta afección. El proveedor de
atención médica llevará a cabo un examen físico. El examen puede
mostrar: Reflejos demasiado exagerados Espasticidad (tener espasmos)
Los exámenes de sangre y orina pueden mostrar niveles altos de ácido
úrico. Una biopsia de piel puede mostrar disminución de los niveles de la
enzima HGP.
- Demasiado ácido úrico puede provocar una hinchazón seudogotosa en
algunas de las articulaciones. En algunos casos, se desarrollan cálculos
renales y vesicales. Las personas con Lesch-Nyhan tienen retraso del
desarrollo motor seguido de movimientos anormales y aumento de los
reflejos. Un rasgo llamativo del síndrome de Lesch-Nyhan es el
comportamiento autodestructivo, lo que incluye morderse las yemas de
los dedos y los labios. Se desconoce la forma como la enfermedad causa
estos problemas.
- El síndrome de Lesch-Nyhan se transmite como un rasgo ligado al sexo, o
un rasgo ligado al cromosoma X, y en su mayoría se presenta en niños
varones. Las personas con este síndrome no tienen o tienen muy poca
cantidad de una enzima llamada hipoxantina guanina
fosforribosiltransferasa (HPRT, por sus siglas en inglés) una sustancia que
el cuerpo necesita para reciclar las purinas. Sin esta, se acumulan en el
cuerpo niveles anormalmente altos de ácido úrico.
- El sindrome de Lesch-Nyhan es un trastorno que se transmite de
padres a hijos (hereditario). Afecta la forma como el cuerpo
produce y descompone las purinas. Las purina son una parte
normal del tejido humano y ayudan a conformar la constitución
genética del cuerpo. También se encuentran en muchos alimentos
diferentes.
- Cockayne
- El síndrome de Cockayne (SC) es una
enfermedad multisistémica
caracterizada por estatura baja,
apariencia facial característica,
envejecimiento prematuro,
fotosensibilidad, disfunción neurológica
progresiva y discapacidad intelectual.
- La edad del paciente en el momento de aparición de la enfermedad y su
gravedad son variables. En la forma clásica del SC (tipo I) los primeros síntomas
aparecen durante el primer año de vida. También se han descrito casos con
síntomas más graves y de inicio temprano (tipo II) y casos en los que la aparición
de la enfermedad es más tardía y que presentan síntomas moderados (tipo III).
Los signos más frecuentes de la enfermedad incluyen: retraso de crecimiento
progresivo, discapacidad intelectual, ataxia cerebelosa, espasticidad, neuropatía
periférica desmielinizante, retinopatía pigmentaria, sordera neurosensorial y
anomalías dentarias (presencia de caries). Los rasgos dismórficos típicos
incluyen: microcefalia, orejas grandes, nariz fina y enoftalmia. En algunos
pacientes se observa también cataratas y fotosensibilidad cutánea. La lipoatrofia
subcutánea está presente y puede conducir a signos de envejecimiento
prematuro en la piel. El síndrome COFS (ver este término) corresponde a la f
- Igual que el xeroderma pigmentoso y la tricotiodistrofia (ver estos términos), el SC
pertenece al grupo de enfermedades que afectan a la reparación del DNA por
escisión de nucleótidos (NER). Las células presentan una alteración específica en la
vía de reparación acoplada con la transcripción (TCR), un subtipo de la NER implicada
en la reparación de las lesiones de DNA inducidas por los rayos UV en los genes
activamente transcritos. Alteraciones adicionales en la transcripción basal o en la
reparación oxidativa también se han propuesto para explicar los síntomas
no-cutáneos del SC. Se han descrito mutaciones en dos genes importantes: ERCC6
(CSB; 10q11) y ERCC8 (CSA; 5q12.1). Hasta el momento, no se ha encontrado ninguna
correlación entre los tres tipos de SC y los genes implicados.
- Métodos diagnósticos El diagnóstico se basa en la detección de la
alteración específica de la vía TCR, que puede ser identificada mediante
el análisis de la recuperación de la síntesis de ARN en fibroblastos en
cultivo, después de la irradiación con rayos UV. Este test de la
reparación del DNA es indispensable para el diagnóstico del SC. La
imaginería cerebral revela hipomielinización difusa de la substancia
blanca, calcificaciones en el putamen y atrofia vermiana.
- El manejo es sintomático e incluye fisioterapia, protección solar, audífonos y frecuentemente,
gastrostomía o alimentación por sonda.
- El manejo es sintomático e incluye fisioterapia, protección solar, audífonos y frecuentemente,
gastrostomía o alimentación por sonda.
- Métodos diagnósticos El diagnóstico se basa en la detección de la
alteración específica de la vía TCR, que puede ser identificada mediante
el análisis de la recuperación de la síntesis de ARN en fibroblastos en
cultivo, después de la irradiación con rayos UV. Este test de la
reparación del DNA es indispensable para el diagnóstico del SC. La
imaginería cerebral revela hipomielinización difusa de la substancia
blanca, calcificaciones en el putamen y atrofia vermiana.
- Igual que el xeroderma pigmentoso y la tricotiodistrofia (ver estos términos), el SC
pertenece al grupo de enfermedades que afectan a la reparación del DNA por
escisión de nucleótidos (NER). Las células presentan una alteración específica en la
vía de reparación acoplada con la transcripción (TCR), un subtipo de la NER implicada
en la reparación de las lesiones de DNA inducidas por los rayos UV en los genes
activamente transcritos. Alteraciones adicionales en la transcripción basal o en la
reparación oxidativa también se han propuesto para explicar los síntomas
no-cutáneos del SC. Se han descrito mutaciones en dos genes importantes: ERCC6
(CSB; 10q11) y ERCC8 (CSA; 5q12.1). Hasta el momento, no se ha encontrado ninguna
correlación entre los tres tipos de SC y los genes implicados.
- La edad del paciente en el momento de aparición de la enfermedad y su
gravedad son variables. En la forma clásica del SC (tipo I) los primeros síntomas
aparecen durante el primer año de vida. También se han descrito casos con
síntomas más graves y de inicio temprano (tipo II) y casos en los que la aparición
de la enfermedad es más tardía y que presentan síntomas moderados (tipo III).
Los signos más frecuentes de la enfermedad incluyen: retraso de crecimiento
progresivo, discapacidad intelectual, ataxia cerebelosa, espasticidad, neuropatía
periférica desmielinizante, retinopatía pigmentaria, sordera neurosensorial y
anomalías dentarias (presencia de caries). Los rasgos dismórficos típicos
incluyen: microcefalia, orejas grandes, nariz fina y enoftalmia. En algunos
pacientes se observa también cataratas y fotosensibilidad cutánea. La lipoatrofia
subcutánea está presente y puede conducir a signos de envejecimiento
prematuro en la piel. El síndrome COFS (ver este término) corresponde a la f
- El síndrome de Cockayne (SC) es una
enfermedad multisistémica
caracterizada por estatura baja,
apariencia facial característica,
envejecimiento prematuro,
fotosensibilidad, disfunción neurológica
progresiva y discapacidad intelectual.
- Moebius
- Consiste en la parálisis congénita, bilateral, de los
nervios craneales VII (facial) y VI (oculomotor externo
o abducens). Con frecuencia asocia parálisis en otros
nervios craneales siendo los más frecuentemente
afectados el hipogloso (XII), vago (X), acústico (VIII) y
glosofaríngeo (IX). También pueden presentarse
asociadas al s. Moebius malformaciones
músculoesquelé- ticas como pies zambos, anomalía
de Poland y defectos de reducción de extremidades
tipo amputación (transverso-terminal). Aunque el
Moebius típico se presenta con parálisis facial
bilateral, la afectación unilateral no lo descarta ya
que hay descritos casos con parálisis facial congénita
unilateral, asociada a parálisis de oculomotor externo
y de otros nervios craneales, así como a
malformaciones músculoesqueléticas típicas del
Moebius (Poland, defectos reducción...).
- Es una enfermedad heterogénea, y las diferentes
causas siguen siendo básicamente desconocidas. No
obstante, numerosas evidencias procedentes de
estudios de necropsias, de observaciones clínicas y de
los actuales estudios de neuroimagen (tomografía axial
computarizada [TAC], resonancia magnética), apoyan la
hipótesis de que casi siempre hay o bien una agenesia
de los núcleos de los nervios craneales VI y VII, o bien
esos núcleos comienzan su desarrollo de forma normal
y posteriormente,
- En resumen, las causas del Moebius serían múltiples (genéticas,
factores teratógenos, anomalías placentarias, etc.) pero en todos
los casos habría un mecanismo común: la agenesia ó destrucción
de los núcleos de los nervios VII y VI situados en tronco cerebral.
- El diagnóstico es clínico y en los primeros meses de vida no
siempre va a ser fácil reconocer un s. de Moebius por lo que en
ocasiones se retrasa el diagnóstico. Las dificultades para la
alimentación, junto a la inexpresividad facial y las
malformaciones asociadas hace que a veces se diagnostique
inicialmente a estos niños como “síndrome polimalformativo”,
“parálisis cerebral” ó “retraso psicomotor”. Dado que la
capacidad intelectual rara vez está afectada esto puede llevar a
estigmatizar al paciente y a condicionar su manejo en los
primeros años de vida
- La falta de movilidad de los músculos de los párpados, que pertenecen al
territorio del Facial, producirá mala humidificación del ojo por la ausencia
de parpadeo, con el consiguiente peligro de desarrollar ulceras corneales
- La afectación del oculomotor externo producirá imposibilidad de
efectuar la mirada lateral y estrabismo convergente.
- La afectación del oculomotor externo producirá imposibilidad de
efectuar la mirada lateral y estrabismo convergente.
- La falta de movilidad de los músculos de los párpados, que pertenecen al
territorio del Facial, producirá mala humidificación del ojo por la ausencia
de parpadeo, con el consiguiente peligro de desarrollar ulceras corneales
- El diagnóstico es clínico y en los primeros meses de vida no
siempre va a ser fácil reconocer un s. de Moebius por lo que en
ocasiones se retrasa el diagnóstico. Las dificultades para la
alimentación, junto a la inexpresividad facial y las
malformaciones asociadas hace que a veces se diagnostique
inicialmente a estos niños como “síndrome polimalformativo”,
“parálisis cerebral” ó “retraso psicomotor”. Dado que la
capacidad intelectual rara vez está afectada esto puede llevar a
estigmatizar al paciente y a condicionar su manejo en los
primeros años de vida
- En resumen, las causas del Moebius serían múltiples (genéticas,
factores teratógenos, anomalías placentarias, etc.) pero en todos
los casos habría un mecanismo común: la agenesia ó destrucción
de los núcleos de los nervios VII y VI situados en tronco cerebral.
- Es una enfermedad heterogénea, y las diferentes
causas siguen siendo básicamente desconocidas. No
obstante, numerosas evidencias procedentes de
estudios de necropsias, de observaciones clínicas y de
los actuales estudios de neuroimagen (tomografía axial
computarizada [TAC], resonancia magnética), apoyan la
hipótesis de que casi siempre hay o bien una agenesia
de los núcleos de los nervios craneales VI y VII, o bien
esos núcleos comienzan su desarrollo de forma normal
y posteriormente,
- Consiste en la parálisis congénita, bilateral, de los
nervios craneales VII (facial) y VI (oculomotor externo
o abducens). Con frecuencia asocia parálisis en otros
nervios craneales siendo los más frecuentemente
afectados el hipogloso (XII), vago (X), acústico (VIII) y
glosofaríngeo (IX). También pueden presentarse
asociadas al s. Moebius malformaciones
músculoesquelé- ticas como pies zambos, anomalía
de Poland y defectos de reducción de extremidades
tipo amputación (transverso-terminal). Aunque el
Moebius típico se presenta con parálisis facial
bilateral, la afectación unilateral no lo descarta ya
que hay descritos casos con parálisis facial congénita
unilateral, asociada a parálisis de oculomotor externo
y de otros nervios craneales, así como a
malformaciones músculoesqueléticas típicas del
Moebius (Poland, defectos reducción...).
- Moebius
- Consiste en la parálisis congénita, bilateral, de los nervios
craneales VII (facial) y VI (oculomotor externo o abducens).
Con frecuencia asocia parálisis en otros nervios craneales
siendo los más frecuentemente afectados el hipogloso
(XII), vago (X), acústico (VIII) y glosofaríngeo (IX). También
pueden presentarse asociadas al s. Moebius
malformaciones músculoesquelé- ticas como pies zambos,
anomalía de Poland y defectos de reducción de
extremidades tipo amputación (transverso-terminal).
Aunque el Moebius típico se presenta con parálisis facial
bilateral, la afectación unilateral no lo descarta ya que hay
descritos casos con parálisis facial congénita unilateral,
asociada a parálisis de oculomotor externo y de otros
nervios craneales, así como a malformaciones
músculoesqueléticas típicas del Moebius (Poland, defectos
reducción...).
- Es una enfermedad heterogénea, y las diferentes causas siguen
siendo básicamente desconocidas. No obstante, numerosas
evidencias procedentes de estudios de necropsias, de
observaciones clínicas y de los actuales estudios de neuroimagen
(tomografía axial computarizada [TAC], resonancia magnética),
apoyan la hipótesis de que casi siempre hay o bien una agenesia
de los núcleos de los nervios craneales VI y VII, o bien esos
núcleos comienzan su desarrollo de forma normal y
posteriormente, en algún momento del desarrollo embriofetal, y
probablemente por algún evento de tipo isquémico, se destruyen.
Experimentos provocando alteraciones en el flujo uterino de
animales de laboratorio han conseguido reproducir los defectos
del Moebius observados en humanos.
- No hay ningún tratamiento curativo del síndrome de Moebius. No
son efectivas ningun tipo de maniobras de estimulación o
rehabilitación facial.
- No hay ningún tratamiento curativo del síndrome de Moebius. No
son efectivas ningun tipo de maniobras de estimulación o
rehabilitación facial.
- Es una enfermedad heterogénea, y las diferentes causas siguen
siendo básicamente desconocidas. No obstante, numerosas
evidencias procedentes de estudios de necropsias, de
observaciones clínicas y de los actuales estudios de neuroimagen
(tomografía axial computarizada [TAC], resonancia magnética),
apoyan la hipótesis de que casi siempre hay o bien una agenesia
de los núcleos de los nervios craneales VI y VII, o bien esos
núcleos comienzan su desarrollo de forma normal y
posteriormente, en algún momento del desarrollo embriofetal, y
probablemente por algún evento de tipo isquémico, se destruyen.
Experimentos provocando alteraciones en el flujo uterino de
animales de laboratorio han conseguido reproducir los defectos
del Moebius observados en humanos.
- Consiste en la parálisis congénita, bilateral, de los nervios
craneales VII (facial) y VI (oculomotor externo o abducens).
Con frecuencia asocia parálisis en otros nervios craneales
siendo los más frecuentemente afectados el hipogloso
(XII), vago (X), acústico (VIII) y glosofaríngeo (IX). También
pueden presentarse asociadas al s. Moebius
malformaciones músculoesquelé- ticas como pies zambos,
anomalía de Poland y defectos de reducción de
extremidades tipo amputación (transverso-terminal).
Aunque el Moebius típico se presenta con parálisis facial
bilateral, la afectación unilateral no lo descarta ya que hay
descritos casos con parálisis facial congénita unilateral,
asociada a parálisis de oculomotor externo y de otros
nervios craneales, así como a malformaciones
músculoesqueléticas típicas del Moebius (Poland, defectos
reducción...).
Medienanhänge
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Möchten Sie kostenlos Ihre eigenen Mindmaps mit GoConqr erstellen? Mehr erfahren.