Beschreibung
|
|
Erstellt von Cher Bachar
vor fast 12 Jahre
|
|
Seite 1
{kind=link}
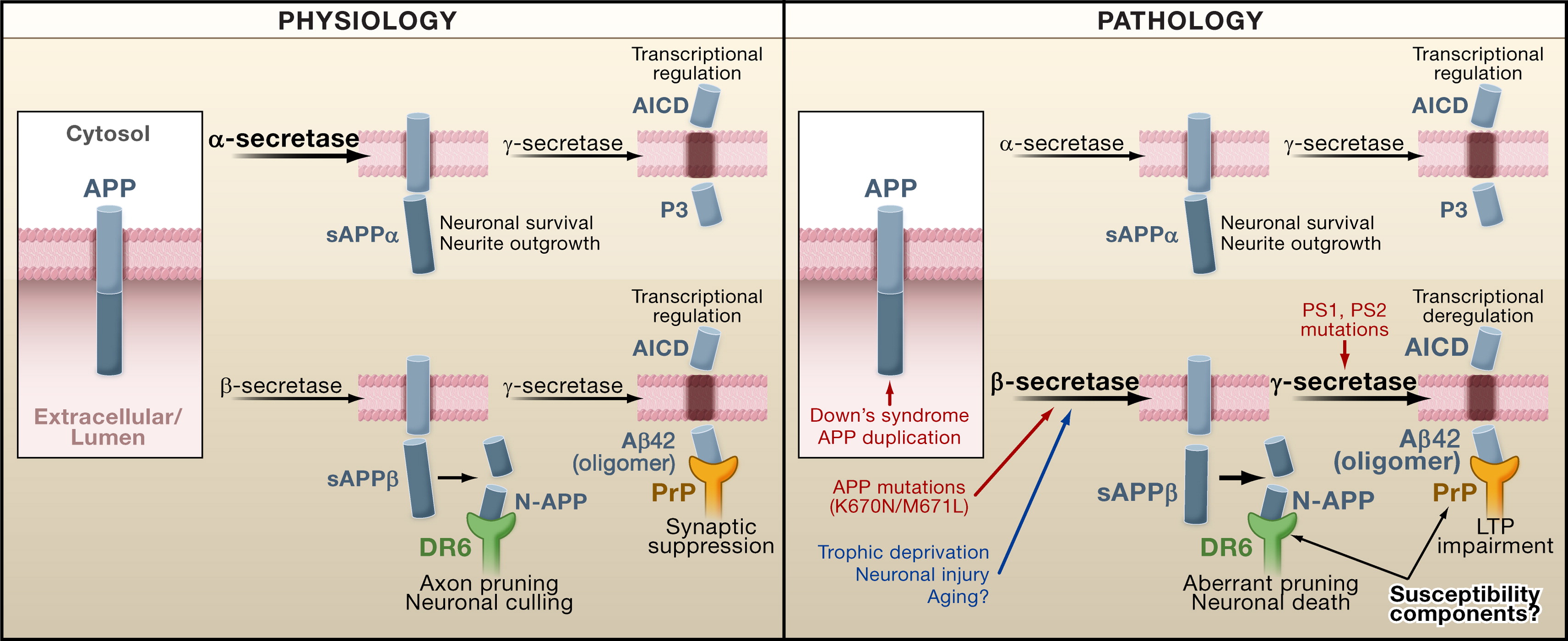
Figure 1. APP Processing in Physiology and Pathology(Left) APP is cleaved by α-secretase or β-secretase, and then by γ-secretase to produce various APP products, including N-terminal APP fragment (N-APP), amyloid-β peptide (Aβ), and APP intracellular domain (AICD). The various fragments perform distinct physiological functions: N-APP induces axonal pruning and neuronal culling by binding to DR6, and Aβ42 oligomers maintain synaptic homeostasis by binding to the prion protein (PrP). (Right) During AD pathogenesis, APP processing is increased or altered by genetic factors such as trisomy 21 in Down's syndrome, mutations in APP, or putative sporadic mechanisms. The resulting increase in Aβ42 and N-APP impairs synaptic plasticity and induces aberrant neuronal and axonal degeneration in a temporal and/or spatial pattern that may be dependent on the expression of the receptors PrP and DR6. Red, genetic factors driving APP processing; blue, possible events inducing sporadic AD; PS1 and PS2, presenilin 1 and 2.
{kind=link}
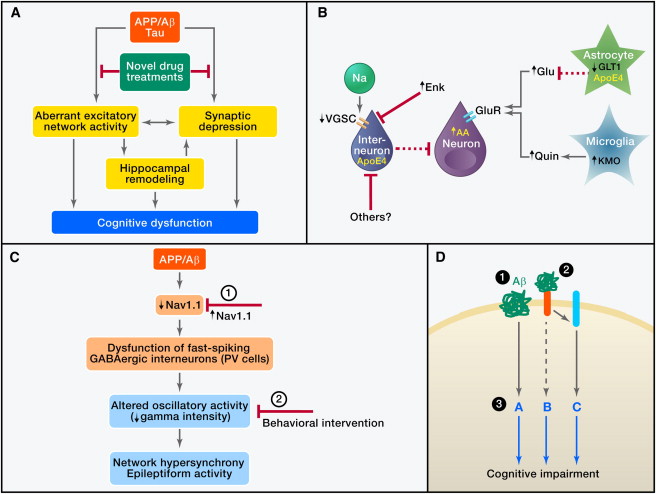
Figure 2. Aβ in AD Pathogenesis and Related Therapeutic Strategies(A) Pathogenic Aβ assemblies cause tau-dependent aberrant excitatory network activity and synaptic depression, which may lead directly, or indirectly through hippocampal remodeling, to cognitive dysfunction. Identification and blockade of the upstream mechanisms that trigger this cascade are important objectives.(B) Aβ-dependent aberrant network activity may be caused by impairments of inhibitory interneurons resulting from depletion of voltage-gated sodium channel subunits (VGSC), intracellular activities of neurotoxic apoE4 fragments, or elevated metenkephalin (Enk) levels. Additional mechanisms that may cause neuronal overexcitation in AD include impairment of glutamate transporters (e.g., GLT1), resulting in decreased clearance of glutamate (Glu), and increased neuronal or microglial production of factors that promote excitotoxicity, including arachidonic acid (AA) and quinolinic acid (Quin).(C) hAPP mice have depletions of the VGSC subunit Nav1.1 that impair the function of parvalbumin (PV)-positive inhibitory interneurons (PV cells), resulting in reduced intensity of gamma oscillations and network hypersynchrony. These network abnormalities can be prevented by increasing Nav1.1 levels in PV cells (1) and counteracted, at least temporarily, by behavioral interventions that enhance gamma activity (2).(D) Aβ may impair neuronal functions by receptor-mediated and receptor-independent mechanisms. Potential therapeutic strategies include (1) lowering Aβ production with secretase inhibitors or modulators and enhancing Aβ clearance with antibodies or activation of Aβ-degrading enzymes, (2) blocking the interaction of Aβ with cell surface receptors, and (3) modulating downstream pathways to make the brain more resistant against whatever Aβ cannot be removed effectively and safely.
{kind=link}
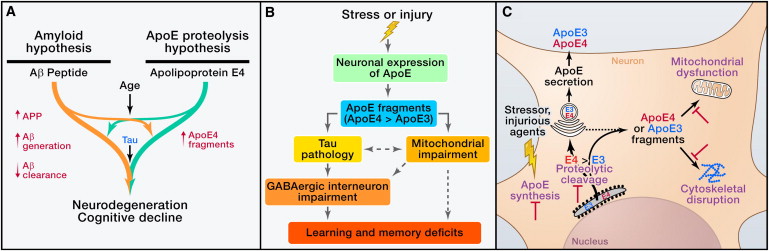
generation of B-amyloid
Aβ in AD Pathogenesis and Related Therapeutic Strategies
apoE
Möchten Sie kostenlos Ihre eigenen Notizen mit GoConqr erstellen? Mehr erfahren.