18924443
GoConqr Rezension
Repaso de Genética
Karteikarten von Eva Rocío Martínez Juanes, aktualisiert more than 1 year ago
|
|
Erstellt von Eva Rocío Martínez Juanes
vor mehr als 5 Jahre
|
|
| Frage | Antworten |
| Factor V Leiden | Resistencia a la proteína C activada. Trombofilia hereditaria + frecuente junto con mutaciones G20210A de la protrombina Sustitución Arg/Glu 506. AD. 3% de la población heterocigota. 25% de las trombosis idiopaticas en pacientes sin riesgo. |
| Poliquistosis renal del adulto | AD. Presentacion adulta. HTA, deterioro de función renal y quistes múltiples en riñon e hígado. |
| Distrofia miotonica de Steinert o tipo I | AD. Cromosoma 19q. Impronta materna. Expansión de tripletes DMPK. Mut. Dinamica - Debilidad extremidades. Predominio distal. Mejoria con repetición del movimiento - Cataratas post. subcapsulares bilaterales. -bloqueos conducción cardiaca -intolerancia a carbohidratos -calvicie frontal -ptosis |
| Otoesclerosis | A.D. - Escotoma en audiograma o cuña de carhart - Rine - / Weber hacia lado afecto / Schawabach +++ - Signo de schwatze. mucosa del promontorio hiperemica en otoscopia. - ausencia de reflejo estapedial. |
| Miocardiopatia hipertrofica hereditaria | AD. - hipertrofia asimétrica - disfunciones diastolica - obstruccionista a la salida del VI -Muerte subida |
| Corea de Hungtinton | AD. Proteina Hungtintina. Repeticiones CAG en gen IT15. Mutacion dinamica (anticipacion) C4p. Impronta paterna. |
| Enfermedad de Von Villebrand | A.D |
| Diabetes MODY | En jóvenes. Similar a tipo II. AD |
| Ataxia espinocerebelosa (SC) | todas AD. Repetición CAG. Dinamica. Gent ATXN1. SCA1. 6p22.3, Impronta paterna. |
| Charcot-Marie- Tooth | Neuropatia hereditaria mas frecuente. - Tipo CMT2. AD. Mutacion MFN 2 (mitofusin 2. Cromosoma 1p. Tipo axonal la + frecuente (45%). - Tipo CMTX1. X recesiva o dominante. Gen GJB1. Fenotipo desmielinizante. |
| Rendu - Osler | Telengectasia hemorragia hereditaria. AD. Epistaxis recurrente. Telangectiasas en lengua y mucosa total. Anemia crónica por sangrado. Empeora con la edad. MAV en pulmón (en estos casos pueden tener poliglobulia) |
| MEN 1 | AD. Sindrome de wermer. Cr11. Tumor neuroendocrino del pancreas+ tumor hipofisario + hiperparatiroidismo |
| MEN 2A | Enf. Sipple. AD. CR10 Gen RET. Carcinoma medular de tiroides + ca paratiroides + feocromocitoma |
| MEN 2B o 3 | AD. Fenotipo marfanoide |
| Esclerosis Tuberosa | Epiloia. AD. Penetrancia completa. Hemangiomas + manchas hipocromas + crisis epilépticas (síndrome de west o Lennox-gastaut) +/- rabomioma cardiaco |
| Von hippel Lindau | AD. Hemangioblastoma retiniano + feocromocitoma + Ca renal |
| Neurofibromatosis tipo I | Von Recklinghausen. AD. Cr 17. Manchas café con leche (+6) Impronta paterna. |
| Neurofibromatosis tipo II | AD. Shwanomas. Impronta materna. |
| Hipercolesterolemia familiar | AD. Cr. 19 (letras) |
| Hipertrigliceridemia familiar | AD |
| Síndrome de Lynch | Cancer de colon hereditario no polipósico. AD. Genes MSH 2, MSH 6, MLH1, PMS 1 y PMS 2. Inestabilidad en microsatélites. Ca colorrectal en colon derecho en 3 o + familiar. 1 de 1º grado. En <50 años. Afectación de dos generaciones. Colectomía subtotal con anastomosis iliorectal. Pobremente diferenciad, mucinoso, cel. En anillo. Reaccionan mal a qt. Bien a Inmunoterapia |
| Cancer de mama y ovario hereditario | AD. BRCA |
| Poliposis colonica familiar | AD. Gen APC.+ 100 pólipos intestinales |
| Esferocitosis hereditaria | AD |
| Porfiria | AD. EXCEPTO Porfiria aguda de DOSS y Porfiria de Günther. Porfiria cutanea tarda Cr1 |
| Protoporfiria eritropoyetica | AD |
| Enfermedad de Marfan | AD |
| Osteogenesis imperfecta | AD |
| Ictiosis vulgar y epidermiolitica | AD |
| Deficit antitrombina III | Trombofilia hereditaria. AD |
| Deficit proteína C +/- Deficit proteína S | Trombofilia hereditaria. AD. Pueden ir individuales o asociadas |
| Fibrosis Quistica | Mucoviscidosis. AR. Gen CFTR. Cr 7. Mutacion Delta-f508. Tripsina inmunorreactiva en neonatos. Test de sudor >60mEq/L de cloruro. |
| Defectos metabólicos enzimaticos | AR EXCEPTO Enfermedad de Fabry y Síndrome de Hunter (X recesos). Galactosemia Cr9. |
| Gaucher | AR. Enf. Metabólico enzimatico. Lipodisis + frecuente. Anemia + trombocitopenia+ hepatOesplenomegalia + lesiones óseas. Deficit enzima betaglucosidasa acida (glucocerebrosidasa) Cr 1. Mut. N370s |
| Fenilcetonuria | AR. Enf. metabólico enzimatica. |
| Hemocromatosis | AR. Gen HFE locus HLA-A6. Cr6p. Mutacion 282Y (caucásicos), variante H63D. Cribdo poblacional con indice de saturación de transferida y ferrita. Cribado familiar de 1º grado, estudio genético. |
| Ataxia de Friedreich | AR. Mutacion dinamica |
| Drepanocitosis | Anemia falciforme. AR o codominante |
| Ataxia telangectasia | AR |
| Talasemia alfa o beta | AR o codominante. Talasemia beta Cr11. |
| Xeroderma pigmentosa | AR |
| Anemia de Fanconi | AR |
| síndrome de bartter | AR |
| Enfermedad de Wilson | AR. Cr. 13 |
| Def. 21 hidroxilasa | AR |
| Decit de alfa1 antitripsina | codominante. Cr.14 |
| Raquitismo vitaminorresistente | Hipofosfatemia. X dominante |
| Síndrome de Alport | X dominante. Lenticono + microhematuria + hipoacusia neurosensorial |
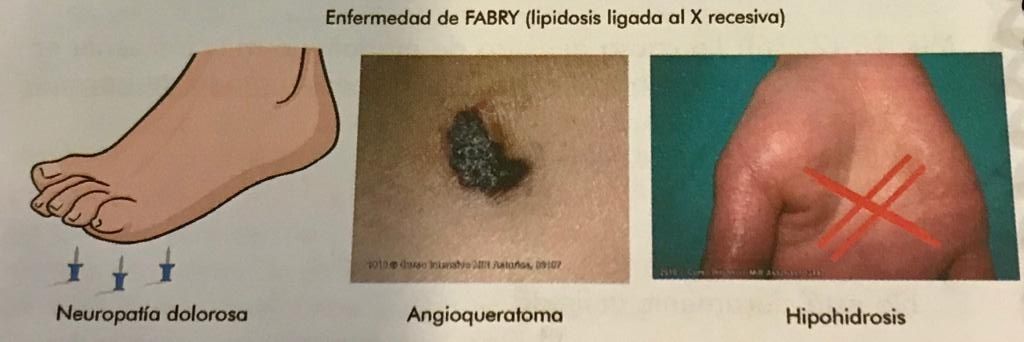
| Enfermedad de Fabry | X recesiva. Enf. metabólico-enzimática. Lipidosis. neuropatia dolorosa + hipodriosis + angioqueratomas |
| Miopatia de Duchene y Becker | X recesiva. Gen de distrofina. |
| Hemofilia A y B | X recesiva |
| Def. Glucosa 6p DHasa | X recesiva. Crisis hemolitica dependientes de habas, antipalúdicos, inf. viral, naftalina (Antipolillas). |
| Enfermedad granulomatosa cronica | X recesiva |
| Inmunodeficiencia Wiskott Aldrich | X recesiva |
| Hipogammaglobulinemia de Bruton | X recesiva. + frac de haemofilus (neumonía, otitis, etc). Recuento global de linfocitos normal. Lin B disminuidos? |
| Síndrome del cromosoma X fragil | X recesivo. Retraso mental, implantación de orejas baja. + predisposición a tumores Repeticion tripletes. Mut. Dinamica |
| Síndrome de Lesch-Nyhan | X recesivo |
| Daltonismo | X recesivo |
| Síndrome de Morris | Feminización testicular. X recesivo |
| Diabetes insípida nefrogenica familiar | X recesiva. Receptor resistente a ADH |
| Neuropatia óptica hereditaria de Leber | ceguera en la juventud que afecta a varones. Herencia mitocondrial. Gen LHON11778a |
| MERF | epilepsia mioclonica de fibras rojas rotas. Herencia mitocondriial |
| MELAS | Síndrome de encefalopatia mitocondria con acidosis láctica y déficits focales que simulan ictus, con fibras rojas rasgadas en la biospsia muscular. Herencia mitocondria |
| Enf. Leigh (NARP) | Neuropatia + ataxia + retinitis pigmentaria. Herencia mitocondrial |
| Sindrome de Pearson | Herencia mitocondrial. Alteracion medular y pancreas exocrino |
| Síndrome de Kearns Sayre | Herencia mitocondrial. Oftalmoplejia +retinitis pigmentaria +/- bloqueo cardiaco +/- alteracion cerebelosa +/- aumento de proteinas LCR |
| Sindrome de Prader-Willi | Impronta paterna. delecion Cr15q11-13. (Delecion de la banda materna?) |
| Sindrome de Angelman | Impronta materna. Cr15. Dos copias materna |
| Distrofia miotónica tipo 2 | PROMM o miopatia proximal miotónica. Expansion trinucleotido gen ZNF9 (tambien llamado CNBP). Dinamica. |
| Tumor de Wilms | Cr11 |
| Hirchsprungd | Cr10 |
| Tumor germinal | Cr.12 |
| Sindrome de Down | Trisomia 21. Canal AV comun. Atresia duodenal. Megacolon aganglionico. Leucemia linfoide. Enfernedad de Alzheimer. Inedtabilidad atlanto axoidea |
| Edwards | Trisomia 18. Mas en mujeres. Retrado mental y ponderoestatural. Hipertonia. Occipucio prominente. Micrognatia. Hipoplasia ungueal.desdos montados. Pie en mecedora o equinovaro. C.I.A y C.I.V. criptorquidea |
| Patau | Trisomia 13. Microcefalia con defectos del cuero cabelludo. Microftalmia, colobomas y cataratas. Cebocefalia. Fisuras labiales y palatinas. Polidactilia postaxial y sindactilia. Malf. SNC y cardiovascular |
| Lejeune | Maullido de Gato. Deleccion brazo corto. Cr 5 p-. Llanto de gato ( |
| Sindrome Li Fraumeni | Mutacion TP53 |
{kind=link}
Möchten Sie mit GoConqr kostenlos Ihre eigenen Karteikarten erstellen? Mehr erfahren.