8397558
Beschreibung
Karteikarten von Amelia Claire, aktualisiert more than 1 year ago
|
|
Erstellt von Amelia Claire
vor mehr als 7 Jahre
|
|
| Frage | Antworten |
| Outline erythrocyte function | erythrocytes (RBCs) contain haemoglobin and transport oxygen around the body to the tissues and bring back CO2 to the alveoli to be expired into the air. Efficacy of oxygen/carbon dioxide transport depends on: Number of erythrocytes (red blood cells, RBCs) in circulation Functional ability of individual erythrocytes |
| outline normal erythrocyte formation and regulation | All blood cells derived from pluripotent stem cells Differentiate to become erythrocytes, platelets or leukocytes when pluripotent cells take the myeloid line and become progenitor cells they are on their way to becoming RBCs RBC formation is regulated by erythropoietin from the kidney which signals the formation of RBCs in the bone marrow, triggered by hypoxia. it takes 3-5 days for RBCs to be formed #recycling haemoglobin# Death of RBCs and phagocytosis by macrophages Iron from haeme recycled via liver to bone marrow for production of new RBCs Other haeme breakdown product biliverdin converted to bilirubin, conjugated in liver and excreted in faeces and urine |
| Define anaemia | a reduction in the number or functionality of circulating RBCs Anaemia can be described by size & number of functional red blood cells & haemoglobin concentration Type confirmed by: * Measuring serum ferritin, iron, vitamin B12 & folic acid * Microscopic examination of blood &/or bone marrow smear |
| explain the terms macrocytic, microcytic, normocytic, hypochromic and normochromic and how they relate to different subtypes/common causes of anaemia | macrocytic -> over large RBC microcytic-> small RBC normocytic-> normal RBC hypochromic -> RBC lacking colour normochromic-> normally coloured RBC microcytic and hypochromic RBCs generally a sign of iron deficiency anaemia microcytic RBCs linked to B12 and folate deficiencies normocytic when linked to anaemia generally linked to chronic illness, renal failure or hemorrhagic anaemia |
| Recognise the key signs symptoms of anaemia and explain how such symptoms relate to the pathophysiology of this condition | fatigue, dizziness, headaches, pale skin, yellowing eyes (haemolytic anaemia) heart palpitations, chest pain, rapid heart rate, low BP, shortness of breath, weakness, pica. enlarged spleen. due to lack of O2 getting to tissues |
| Explain the pathophysiology of and treatments for: Iron deficiency anaemia | pathophysiology: microcytic and hypocrhomic RBCs poor iron intake or absorption increase in iron requirements (pregnancy) chronic blood loss (e.g. ulcer) this leads to reduced production of haemoglobin, reduced production of erythrocytes RBCs product contain less haemoglobin and are smaller to maintain haemoglobin concentration. treat with iron supplements |
| Explain the pathophysiology of and treatments for: Vitamin B12 and/or folic acid deficiency anaemia | B12 is required for methylation of DNA and folic acid is required for synthesis of DNA generally due to poor dietary intake (vegan) treat with B12 and Folic Acid supplements |
| Explain the pathophysiology of and treatments for: Anaemia of chronic renal disease | In severe renal impairment or failure, the kidney fails to produce sufficient erythropoietin. hence, RBC count is reduced. The size and colour of the RBCs are normal. Haemolysis may occur due to dialysis. Dialysis also removes Folate from blood. to treat use Epo Agonists: rhEpo - epoietin alfa and beta half life 6-9 hours IV or subcut synthetic erythropoietin (darbepoietin alfa) two additional carbohydrate chains, increased half life of 25 hours IV or subcut |
| Describe the anatomy of the heart | Chambers of the Heart 4 chambers: – upper 2 atria – lower 2 ventricles • Atria receive blood into heart • Muscles of atria and ventricles separated by connective tissue “skeleton” Right(R) and left(L) atria separated by inter‐ atrial septum • Septum has fossa ovalis – Shallow depression in wall where foramen ovale of foetal heart was • closes over at birth • Auricle=“flap”of expandable atrium visible when not filled Right ventricle pumps blood to pulmonary circulation– Thinner wall, – Less pressure than left ventricle, Left Ventricle pumps blood to systemic circulation – Thicker wall – Greater pressure – Rounder in shape |
| Pulmonary circuit Systemic circuit | • Pulmonary circuit – Carries blood to and from gas exchange surfaces of lungs • Systemic circuit – Carries blood to and from the rest of body |
| Identify the layers of the heart wall | 1.Epicardium (visceral layer of serous pericardium) 2.Myocardium (cardiac muscle tissue) • Bulk of heart tissue • Provides pumping action 3. Endocardium • Continuous with endothelial lining of great vessels |
| Describe valves of the heart | Direct blood flow through and out of the heart • Valves prevent back flow of blood • Composed of dense connective tissue covered by endothelium AV VALVES Lie between atria and ventricles – rightAV‐tricuspid – left AV ‐ bicuspid (mitral) • Cusps linked by chordae tendinae to papillary muscle • Pressure in atria > pressure in ventricles – valves will open and blood flows into ventricles • Pressure in ventricles > pressure in atria – Valves close and blood flows into aorta or PA Semilunar Valves Between ventricles & major arteries (aorta & PA) • Prevent blood flowing back into heart • Pulmonary semilunar valve – At base of pulmonary arterial trunk • Aortic semilunar valve – Atbaseofaorta • No valves between veins and atria |
| Describe the blood flow through the heart | Right atrium receives blood from systemic circulation via – Superior vena cava (SVC) – Inferior vena cava (IVC) – Coronary sinus • Right atrium to Right Ventricle through AV valve • Right ventricle to pulmonary trunk & pulmonary arteries • Then into pulmonary circulation Blood passes through lungs and returns to heart through 4 pulmonary veins • Pulmonary veins into Left atrium • Left atrium to Left ventricle through AV valve • Then into systemic circulation via aorta. |
| Describe the coronary circulation | .• Coronary arteries – branch from ascending aorta – fill upon diastole – carry oxygenated blood to the myocardium • Pulsatile blood flow • Little flow during systole Coronary Sinus carries deoxygenated blood back to right atrium |
| Describe the conducting system of the heart | comprised of AV bundle • Right and left bundle branches • Purkinje fibres Sinoatrial (SA) Node (pacemaker) – Spontaneously depolarises (80‐100 times/min) • Rate of depolarisation modified by neurotransmitters from ANS • E.g. resting HR slower than rate of depolarisation of SA node due to parasympathetic tone (constant low level of vagal input) • Atrioventricular (AV) node – Spontaneous depolarisation 40‐60 times/min – Conduction slows atAV node(AV nodal delay) • Delay allows atria time to contract |
| • Describe a cardiac action potential | Resting membrane potential = – 90mV • Excitation (neighbouring cell)MP toward firing threshold (~‐ 75mV)action potential ‐ three phases: 1. Rapid Depolarisation • Voltage‐gated sodium channels open • Rapid influx of Na+ 2. Plateau • Na+ channels close rapidly (~+30mV) Na+ efflux • Voltage‐gated slow Ca2+ channels open (slowly for long time) Ca2+ influx • Balances slow Na+ outflow (~0mV) 3. Repolarisation • Voltage‐gated slow Ca2+ channels close • Voltage‐gated slow K+ channels openK+ efflux |
| Explain the electrical and mechanical events of the cardiac cycle | 1. SA node mass of cells (pacemaker) – spontaneously generates an AP 2. Stimulus spreads across atria & reaches AV node 3. AV nodal delay~100msec – Atrial contraction begins 4. AP spreads along AV bundle, bundle fibres and Purkinje fibres 5. AP relayed across ventricles – muscle of ventricles contract Cardiac cycle = The period between the start of one heartbeat and the beginning of the next – Includes both contraction and relaxation Cardiac cycle • Phases of the Cardiac Cycle – Within any one chamber • Systole(contraction) • Diastole(relaxation) • For HR 75 bpm – Ventricular Systole =270 msec – Ventricular Diastole = 530 msec |
| Define cardiodynamic variables | • P wave ‐ atrial depolarisation • PR segment ‐ AV nodal delay; atria contracted and emptying • QRS wave ‐ ventricular depolarisation • ST segment ‐ ventricles contracting and emptying • T wave ‐ ventricular repolarisation • TP segment ‐ heart at rest and ventricles (and atria) are filling |
| Explain the factors affecting cardiac output, HR and stroke volume | 1. Autonomic Innervation of the heart – Both SNS and PNS innervate the SA and AV nodes and atrial & ventricular muscle cells – In ventricles SNS>>>>PNS – Controlled by cardiac centres in medulla oblongata • Cardio acceleratory centre(SNS;HR) • Cardio inhibitory centre(PNS;HR) – Reflex pathways regulate cardiac centres,e.g. baroreceptors (BP) & chemoreceptors (CO2 and O2) & higher order CNS Autonomictone – Healthy, resting heart PNS activity > SNS activity Resting HR (70‐80bpm) < SA node depolarisation (~100bpm) – Autonomic tone & dual innervationdelicate adjustments to meet demands • Effects on the SA node – ANS changes rate of spontaneous depolarisation or duration of repolarisation alters HR by changing time required for cells to reach threshold Hormones Adrenaline, noradrenaline & thyroid hormones HR (SA node) – Adrenaline also affects contractile cells * Atrial reflex Adjustments to HR depending on venous return (amount of blood returning to heart through veins) – Venous return – Directly affects nodal cell smore rapid depolarisation; HR |
| Describe the structure and function of the 5 types of blood vessels | Five types of blood vessels: 1. Arteries 2. Arterioles 3. Capillaries 4. Venules 5. Veins • Larger blood vessels served by own blood vessels located within their walls structure supports functions |
| Explain the mechanisms that regulate blood flow through arteries, capillaries and veins | haemodynamics Blood flow – volume of blood that flows through a tissue per unit time – determined by blood pressure and resistance • Blood Flow is: – proportional to the pressure gradient – Inversely proportional to resistance (Opposition to blood flow due to friction between blood and vessel wall) |
| Discuss the movement of fluid between capillaries and interstitial fluid | Substances enter and leave capillaries by three methods: 1. diffusion (most important) 2. transcytosis (vesicular transport) 3. bulk flow (filtration and absorption) |
| List the roles of cholesterol within the body | 3 primary functions: Component for cell membranes - ‘building block’ for tissues Precursor for steroid hormones - including the sex hormones Required for production of bile acids - essential in digestion of fats Cholesterol is also involved in: transport of essential fatty acids insulation of nerves Precursor for vitamin D production |
| Describe the classification and characteristics of: TGs, chylomicrons, VLDL | Chylomicrons • largest lipoprotein, diameter 100-1000 nm • lowest density due to high lipid:protein ratio • Highest triglyceride content • Contain apoprotein B-48 • Transport triglycerides & cholesterol from diet in gastrointestinal tract to tissues VLDL • Very low density lipoprotein, precursor for LDL • 2nd largest lipoprotein, diameter 30-80 nm • 2nd highest % triglyceride content • Contain apoprotein B-100 • Transport cholesterol & synthesized triglycerides to tissues |
| Describe the classification and characteristics of: LDL and HDL | LDL • Low density lipoprotein • Diameter 20-30 nm • Highest in cholesterol esters as a % of weight • Contains apoprotein B-100 • Some take up by tissues, some by liver • When taken up by tissue can lead to atherosclerosis HDL • High density lipoprotein • Diameter 7-20 nm • Highest density due to low lipid:protein ratio • Contain apoproteins A1 and A2 • Absorb cholesterol from tissue breakdown & transfer it to VLDL & LDL |
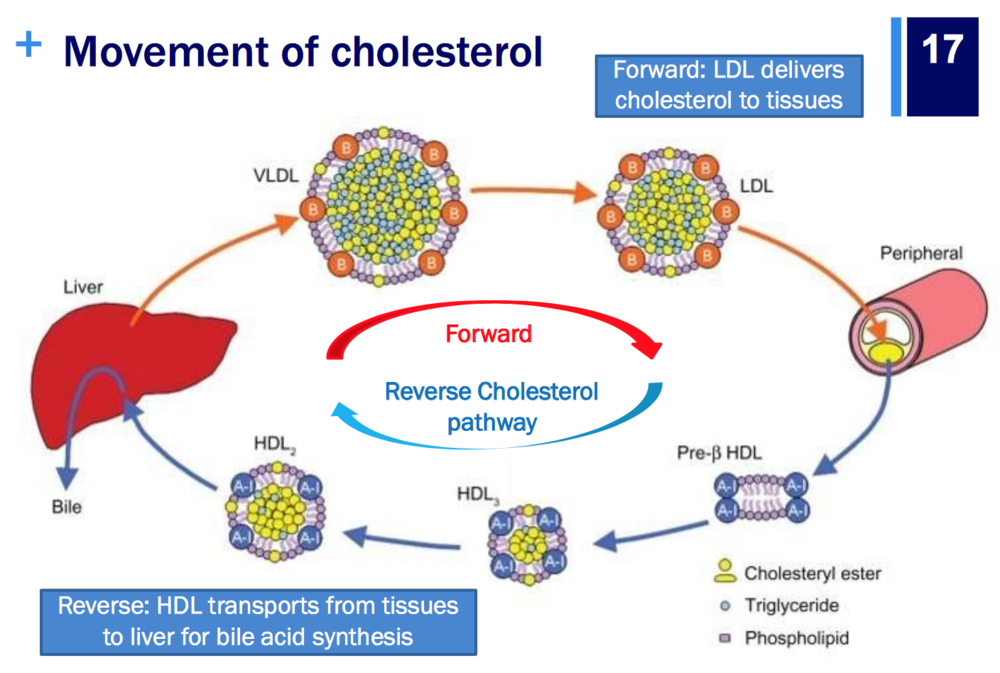
| Explain the endogenous & exogenous cholesterol sources & pathways including the ‘reverse transport’ of cholesterol liver makes cholesterol (de novo) - (endogenous) and we consume it in non plant based food in our diets (exogenous) | |
| Describe the etiology of primary & secondary dyslipidaemia | Primary dyslipidemia Caused by genetic defects in lipoprotein metabolism Secondary dyslipidemia Attributable to another cause, for example: Medical conditions Lifestyle factors Sedentary lifestyle, cigarette smoking, excessive saturated & trans-fatty acid consumption Medications Isotretinoin, tamoxifen, cyclosporin 20 |
| List the physical symptoms of dyslipidaemia | Dyslipidaemia is primarily asymptomatic however: Potential physical findings include: xanthomas (cholesterol deposits in tissue) pancreatitis (inflammation of pancreas) renal or hepatic disease peripheral vascular disease cerebrovascular disease coronary artery disease |
| Describe the mechanism of action, common adverse effects and interactions of drugs used to treat dyslipidaemia: statins | *statins are HMG-CoA reductase inhibitors. *HMG-CoA reductase is the rate limiting enzyme in the mevalonate pathway (which produces cholesterol) 1)as HMG-CoA reductase is inhibited, there is a decreased concentration of cholesterol in the cell 2) low intracellular cholesterol stimulates LDL receptor synthesis 3) increased LDL receptors promotes uptake of LDL into cell from blood 4) low intracellular cholesterol reduces the secretion of VLDLs into blood Adverse Effects: GIT, headache, liver function, muscle issues (myalgia, myositis, most seriously Rhabdomyolysis) Interactions: CYP450 drug interactions |
| Describe the mechanism of action, common adverse effects and interactions of drugs used to treat dyslipidaemia: fibrates | Fenofibrate & gemfibrozil ‘Fibr’ in the name = fibrate Activate peroxisome proliferator-activated receptors (PPAR) & modulate lipoprotein synthesis & catabolism PPAR alters expression of lipid metabolism genes MOA increase transcription of the genes for lipoprotein lipase and apoproteins apoA1 and apoA5 (ligands for specific receptors) fibrates increase uptake of LDLs by the receptors. PPARα activation = ↑ lipoprotein lipase in muscle ↑ Uptake & oxidation of fatty acid in muscle cells ↑ Fatty acid oxidation in liver ↓ Triglyceride (TG) synthesis ↑ Apoprotein A1 & 2 synthesis, thus increases HDL |
| Describe the mechanism of action, common adverse effects and interactions of drugs used to treat dyslipidaemia: ezetimibe, | site of action - intestinal lumen cholesterol absorption inhibitor blocks sterol carrier protein, hence reduces amount of biliary and dietary cholesterol delivers to lier via chylomicrons. decreases liver's cholesterol store, in creases hepatic absorption of LDLs ,thus increases clearance of LDL from plasma. NPC1L1 protein in cell membrane binds to cholesterol for absorption *** Ezetimibe reduces binding of AP2 clathrin to NPC1L1 to ↓ cholesterol absorption reducing the capacity of AP2 clathrin complex binding to NPC1L1 to facilitate endocytosis of cholesterol Inhibits intestinal and biliary cholesterol absorption → depletes hepatic cholesterol pool → increases expression of the LDL receptor on hepatocytes → increases LDL uptake from blood stream Doesn’t interfere with absorption of triglycerides, fat-soluble vitamins or fat-soluble drugs Metabolised in liver, excreted in bile to GI lumen where it can work again → enterohepatic circuit increases half-life |
| Describe the mechanism of action, common adverse effects and interactions of drugs used to treat dyslipidaemia: bile acid binding resins, | Cholesterol is converted to bile acids in the liver Bile acids are secreted into the GI tract, can be reabsorbed Bile acid binding resins sequester bile in GI tract = ↑ excretion in faeces More LDL cholesterol moves from blood stream to liver to form bile acids |
| Describe the mechanism of action, common adverse effects and interactions of drugs used to treat dyslipidaemia: niacin | Mechanism of action is unclear Thought to decrease VLDL, ILD, LDL by: * Suppressing fatty acid release from adipose tissue *) Reducing TG synthesis, via inhibition of DGAT2 enzyme *) Increasing apoprotein B catabolism Thought to increase HDL by: *)Increasing hepatic uptake of & production of apoprotein A * Reducing cholesteryl ester transfer protein * Reducing HDL uptake by inhibiting beta chain ATP synthase |
| Describe the mechanism of action, common adverse effects and interactions of drugs used to treat dyslipidaemia: PCSK9 inhibitors | Humanised monoclonal antibodies that bind to PCSK9 Increase the livers ability to remove LDL from plasma First approved in Aust 2015 alirocumab & evolocumab Given subcut every 2 - 4 weeks Reduce LDL by 50-60%, regardless of other treatments being used PCSK9 promotes lysosomal degradation of the LDL receptor = ↓ amount of LDL receptor on hepatocyte cell membrane MOA: PCSK9 inhibitors block lysosomal degradation of LDL receptor = ↑ amount of LDL receptor on hepatocyte cell membrane = ↑ LDL uptake & ↓plasma LDL |
| Describe the mechanism of action, common adverse effects and interactions of drugs used to treat dyslipidaemia: omega 3 fatty acids | Fish oil is rich in polyunsaturated fatty acids: Eicosapentaenoic acid (EPA) & docosahexaenoic acid (DHA) Sourced from oily fish (e.g. salmon, herring, mackerel) Inhibit release of TGs from the liver Reduces VLDL and therefore LDL Stimulate lipoprotein lipase to increase clearance of TGs from plasma May have anticoagulant and anti-inflammatory effects Adverse effects: gastrointestinal upset, fishy aftertaste |
| what is atherosclerosis | The hardening of an artery, characterized by atheromatous plaque formation in the inner layer of the arteries |
| define: Atheroma Arteriosclerosis Arteriolosclerosis Atherogenic Atherogenesis | Arteriosclerosis: General term describing and hardening (loss of elasticity) of medium or large arteries Atheroma: an abnormal fatty deposit in an artery Arteriolosclerosis: General term describing and hardening (loss of elasticity) of arterioles (small arteries) Atherogenic: Describes substances or processes that cause atherosclerosis Atherogenesis: The developmental process of atheromatous plaques |
| Define: Acute coronary syndrome Coronary artery disease Coronary ischaemic disease | Acute coronary syndrome: Refers to any group of clinical symptoms compatible with acute myocardial ischemia. Coronary artery disease: The most common type of heart disease. Results from atherosclerosis in the coronary arteries. Coronary ischaemic disease: The most common type of heart disease. Results from atherosclerosis in the coronary arteries. |
| Explain the link between cholesterol, atherosclerosis & heart disease | Dyslipidaemia -> Excess LDL accumulates in the intima, is modified to initiate atheroma. Atherosclerosis develops and progresses: |
| Recognise the risk factors for atherosclerosis | Modifiable Dyslipidaemia High BP (↑ permeability vessel wall to LDL) Diabetes mellitus (↑VLDL production, ↑glycation of LDL, associated endothelial dysfunction) Obesity (Contributes to dyslipidaemia, high blood pressure & insulin resistance) Physical inactivity (Contributes to dyslipidaemia, high blood pressure & insulin resistance) Tobacco smoking (↑LDL oxidation, endothelial dysfunction, leukocyte adhesion molecules) Age Plaque build-up takes time to reach critical point Male gender (Lack of estrogen [protective as ↑HDL & ↓LDL]) Hereditary Many genetic factors can be involved |
| Describe the process of plaque formation | Stages in development of atherosclerotic disease: (endothelial injury) Fatty streak development Plaque progression Plaque disruption 1) LDL deposits cholesterol in vessel wall 2a) macrophages engulf cholesterol deposited in vessel wall and become foam cells. 2b) a fatty streak develops between layers of vessel wall 3a) foam cells continue to expand to the core of the plaque 3b) a fibrous outer cap forms from smooth muscle cells and other elements 4a) this is the large unstable plaque with fibrous cap 4b) plaque may rupture 5a) blood clot forms at site of plaque rupture 5b) initially consequences depend where the vessel or artery is located in the body |
| Describe the development of unstable fibrous plaques | a fibrous outer cap forms from smooth muscle cells and other elements |
| Identify the consequences of atherosclerosis occurring in different vessels around the body | could be stroke, heart attack, depending on where the vessel is located |
| What is Haemostasis | Haemostasis is the normal physiological process that stops bleeding, to prevent significant blood loss after vascular injury. |
| What is meant by the vascular phase in haemostasis. | vascular phase: vessel spasm. damaged vessels constrict to reduce local blood flow, minimising blood loss and allowing for haemostatic plug to form. it is the first response to vessel injury and is rapid and transient. |
| explain primary haemostasis | primary haemostasis: = platelet phase adhesion: glycoprotein IIB and von Willebrand Factor (vWF) interact with collagen to help platelets adhere to vessel wall. vWF (like a glue) is secreted by the injured epithelium and helps platelets and cells mesh together. platelet activation: collagen and other factors trigger platelets activation; platelets change shape and become spherical with long dendritic extensions, facilitating adhesion to exposed collagen. This attachment triggers release of ADP and THROMBOXANE A2 (TXA2) - released from platelet cytoplasm, activates nearby platelets and TXA2 causes further vasoconstriction. Platelet aggregation: platelet glycoprotein receptors mediate adhesion to sub endothelial tissue outer surface of platelets become sticky. |
| explain secondary haemostasis | secondary haemostats is the coagulation phase. the platelet plug is relatively unstable and must be reinforced with fibrin to allow time for healing. cascade of enzymatic coagulation reactions that lead to conversion of soluble fibrinogen to insoluble fibrin form a stable fibrin clot. clotting factors present in blood in the inactive form; clotting cascade provides amplification mechanism (activation of one factor catalyses activation of a larger amount of the next factor) coagulating factors named by Roman Numerals. anticoagulant factors are equally as important in this process; ensuring we do not develop thrombi |
| The coagulation cascade - final common pathway in hemostasis | X -> Xa Va Prothrombin (II) -> Thrombin (IIa) Fibrinogen (I) -> Fibrin (Ia) XIIIa cross linked fibrin clot |
| Fibrinolytic Phase of Haemostaisis (thrombis removal) | 1. Tissue plasminogen activator (tPA) released by endothelial cells, binds to plasminogen on fibrin surface 2. Plasminogen in thrombus converted to proteolytic enzyme plasmin 3. Fibrinolysis occurs and thrombus dissolves |
| anticoagulation factors | Tissue Factor Pathway Inhibitor: Produced by endothelial cells, inhibits tissue factor pathway coagulation (extrinsic) Prostacyclin (PGE2): Inhibits action of TXA2 to inhibit platelet aggregation Protein C: Vitamin K dependent, activated by thrombin, inactivates factors Va & VIIa Protein S: Vitamin K dependent, cofactor in activation of protein C, inhibits factor Va-Xa complex Antithrombin: Inactivates factors IIa (thrombin), IXa, Xa, XIa, XIIa, triggers prostacyclin production |
| Recognise factors that contribute to thrombosis & the different clinical presentations for thrombi around the body | thrombosis is the pathological formation of a haemostatic plug within the vasculature in the absence of bleeding. contributing factors: Virchow's Triad hyper-coagulable state vascular wall injury circulatory stasis leg - DVT heart - MI pulmonary - PE carotid - IS |
| arterial vs venous thrombi | arterial: Triggered by damage to the endothelium ‘White’ thrombus, consists of platelets in fibrin meshwork Common causes: • Ruptured atherosclerotic plaques • High blood pressure • Turbulent blood flow Venous: Triggered by stasis or changes in coagulability of blood 'Red’ thrombus, consists of small ‘white’ platelet head with large jelly-like red tail Common causes: • Immobility • Inherited disorders • Certain cancers • Pregnancy |
| Explain the process of fibrinolysis | 1. Tissue plasminogen activator (tPA) released by endothelial cells, binds to plasminogen on fibrin surface 2. Plasminogen in thrombus converted to proteolytic enzyme plasmin 3. Fibrinolysis occurs and thrombus dissolves |
| Describe the mechanism of action, common adverse effects and interactions of antiplatelet drugs: COX inhibitors (aspirin) | .Irreversibly inhibits COX I & II Reduces production of thromboxane A2 (TXA2) to reduced platelet activation Covalently binds COX near active site No DNA in platelets, thus cannot produce more COX, TXA2 production terminated for life of platelet (7-10 days) common AEs Non-selective COX I & II inhibitors can cause adverse effects via reduced production of off-target prostaglandins - (protective prostaglandins COX I, involved with Protect stomach mucosa Promotes platelet aggregation Increase renal perfusion) |
| Describe the mechanism of action, common adverse effects and interactions of antiplatelet drugs: P2Y12 antagonists (clopidogrel, prasugrel, ticagrelor) | Non-competitively inhibit ADP dependent platelet activation & aggregation clopidogrel and prasugrel are taken as pro-drugs and must go through CYP450 to be metabolised into active metabolites. |
| Describe the mechanism of action, common adverse effects and interactions of antiplatelet drugs, including: phosphodiesterase inhibitors (dipyridamole) | Decreases platelet activation by: Inhibiting PDE to increase cAMP within platelets Inhibiting reuptake of adenosine Weak antiplatelet effect Vasodilator properties Headache can limit tolerability Hypotension adverse effect |
| Describe the mechanism of action, common adverse effects and interactions of antiplatelet drugs: glycoprotein IIb/IIIa inhibitors (abciximab, tirofiban) | Occupies glycoprotein IIb/IIIa receptor to inhibit fibrinogen binding, preventing platelet aggregation Abciximab = monoclonal antibody Tirofiban = non-peptide antagonist |
| Explain the intrinsic coagulation pathway and the laboratory tests used to monitor it | Intrinsic Pathway: XII -> XIIa XII->XIIa IX-> IXa VIIIa to common pathway X -> Xa Va Prothrombin (II) -> Thrombin (IIa) Fibrinogen (I) -> Fibrin (Ia) XIIIa cross linked fibrin clot Intrinsic pathway: Activated partial thromboplastin time (aPTT) |
| Explain the extrinsic coagulation pathway and the laboratory tests used to monitor it. | Extrinsic pathway: Prothrombin time (PT) extrinsic pathway VII -> VIIa tissue factor to common pathway X -> Xa Va Prothrombin (II) -> Thrombin (IIa) Fibrinogen (I) -> Fibrin (Ia) XIIIa cross linked fibrin clot International normalised ratio (INR) = Based on PT, ratio of time to clot for warfarinised vs. ‘healthy’ blood Standardises PT across test conditions INR = Prothrombin at Time patient/ Prothrombin at time of Reference Plasma (2-3??) |
| Describe the mechanism of action, common adverse effects and interactions of anticoagulant drugs Including: Vitamin K antagonists (warfarin), | inhibits vitamin K epoxides reductase: *Reduced form of vitamin K required for synthesis of clotting factors II, VII, IX, X *Warfarin inhibits vitamin K epoxide reductase, enzyme that reduces vitamin K *Decreases production of clotting factors II, VII, IX, X interferes with coagulation cascade Delayed onset of action Affects production only, no affect on circulating factors Onset dependent on half-life of factors: Factor VII - 6 h Factor IX - 24 h Factor X - 36 h Factor II - 50 h Also inhibits production of anticoagulant factors Protein C & Protein S Racemic mix: S-warfarin 2-5x more anticoagulant Narrow therapeutic index Susceptible to drug-drug & drug-food interactions |
| Describe the mechanism of action, common adverse effects and interactions of anticoagulant drugs; indirect thrombin inhibitors (heparin, enoxaparin, fondaparinaux) | indirect thrombin inhibitors Induce conformational change to increase in antithrombin III to its efficacy inhibiting Xa (and thrombin for heparin). |
| Describe the mechanism of action, common adverse effects and interactions of anticoagulant drugs; direct thrombin inhibitors (dabigatran) | Reversible, competitive inhibitor of thrombin → prevents conversion of fibrinogen to fibrin: *First oral anticoagulant to enter the market in Australia since warfarin * Does not require INR monitoring & dose adjustments * Less susceptible vs warfarin to drug interactions, but is a P-glycoprotein (P-gp) substrate Very low bioavailability Given as prodrug to increase GIT absorption Hydrolysed to activate in liver Predominately renally cleared |
| Describe the mechanism of action, common adverse effects and interactions of anticoagulant drugs; factor Xa inhibitors (rivaroxaban, apixaban) | * Inhibit Factor Xa to prevent conversion of prothrombin (factor II) to thrombin * Both metabolized CYP3A4 & both substrates of P-gp --> risk of of drug interactions * INR monitoring not useful as no way to relate prothrombin time to efficacy or safety |
| Recognise the factors that may influence INR during warfarin therapy | . |
| Describe the mechanism of action, common adverse effects and interactions of fibrinolytic drugs | put simply: Fibrinolytic agent converts plasminogen into plasmin. Plasmin digests or dissolves fibrin meshwork of clot Clot dissolves First fibrinolytic → streptokinase (SK) n Forms complex with plasminogen to release plasmin * Not specific for clot-bound fibrin * Antibodies generated can block activity if re-administered (or post strep infection) ** Urokinase * Isolated from human urine * Catalyses plasminogen to plasmin * Also not specific for clot bound fibrin |
| Tissue plasminogen activator (t-PA) analogues alteplase & tenecteplase | Analogues of human tPA developed * More specific to clot-bound plasminogen, no immune response ** Alteplase; recombinant t-PA (rt-PA) * Exact copy of human t-PA * Short half-life, need IV infusion ** Tenecteplase *Higher binding affinity for plasmin vs alteplase * Longer half-life, can give IV bolus * Not inactivated by plasminogen acGvator inhibitor (PAI-1) AEs Alter haemostasis more profoundly than any other anticoagulants → Greatest risk for bleeding complications * Many contraindicaGons to fibrinolysis * Concurrent anGplatelet or anticoagulant drugs increase risk of bleeding but combinations can be required |
| Explain purpose of & regulators involved in the fibrinolytic cascade | Activated along side the coagulation cascade * Plasminogen converted to plasmin by tissue plasminogen activator (tPA) & other activators * Regulated by plasminogen activator inhibitors (PAI) & α2-antiplasmin |
| Haemostatic and Reversal Agents | Classes: haemostatic agents Drug-specific reversal agents blood products drugs: HA -> tranexamic acid vitamin K DSRA -> vitamin K Protamine Idarucizumab BP -> Prothrombinex® |
| Describe the mechanism of action, common adverse effects and interactions of reversal agents: tranexamic acid | Inhibits fibrinolysis: * Competitive inhibitor of plasminogen activation * At high doses = non-competitive inhibitor of plasmin * Onset of acGon 5-15 min * Cleared renally – may require dose adjustment |
| Describe the mechanism of action, common adverse effects and interactions of reversal agents: vitamin K | Supplements vitamin K cycle to allow increased production of clotting factors II, VII, IX, X in liver * IV reverses warfarin in 4-6 h *Oral reverses warfarin over 24 h |
| Describe the mechanism of action, common adverse effects and interactions of reversal agents: protamine | Derived from fish sperm * Most effective for heparin * Reverses 60% enoxaparin effect * No reversal of fondaparinux * Weak anticoagulant effect when given alone * When sulfate form given with heparin: *Complex forms → dissociates heparin-antithrombin complex → reduces anticoagulant effect of heparin |
| Describe the mechanism of action, common adverse effects and interactions of reversal agents: idarucizumab | Humanised monoclonal antibody fragment * Binds to dabigatran * Forms stable inactivatesve complex * Reverses anticoagulant effect * Also neutralises metabolites * Immediate effect < 5 min n No direct on haemostasis |
| Recognise situations where reversal of an antiplatelet or anticoagulation medication may be required | . |
| NOAC reversal agents (novel) | Agents under development * Andexanet-alfa (And-α) * Modified recombinant factor Xa, binds to & inactivates Xa inhibitors (apixaban & rivaroxaban) * Ciraparantag * Small molecule, competitively binds to & inactivates all NOACs & indirect thrombin inhibitors |
| what do you give to reverse warfarin | Supplements vitamin K cycle to allow increased production of clotting factors II, VII, IX, X in liver * IV reverses warfarin in 4-6 h *Oral reverses warfarin over 24 h Prothrmobinex-VF |
| what do you give to reverse enoxoparin? | protamine - about 60% effective |
| what do you give to reverse heparin? | protamine When sulfate form given with heparin: *Complex forms → dissociates heparin-antithrombin complex → reduces anticoagulant effect of heparin |
| Explain the role of blood products in managing complications of antiplatelet & anticoagulant drugs | human factors from blood donations are important particularly in haemophilia etc they make factor concentrates: Biostate: Factor VIII & vWB -> used for haemophilia A MonoFIX-VF : Factor IX -> used for haemophilia B Prothrmobinex-VF: Factors II, VII, IX, X. used for II or X deficient and warfarin reversal |
| Recognise situations where reversal of an antiplatelet or anticoagulation medication may be required | if someone is bleeding out or they have had too much anticoagulation medicine - > e.g. if they have a haemorrhage |
| Haemophilia A vs Haemophilia B | Haemophilia A lack of clotting factor VIII Haemophilia B lack of clotting factor IX |
| Explain the pathophysiology of IHD (angina & myocardial infarction) | . |
| Recognise the importance of the local heart vasculature and the factors that decrease myocardial oxygen supply | . |
| Unstable Angina | Unstable angina * Progressive form of angina * Acceleration of stable angina * occurs more frequently & becomes more severe over time * occurs with lesser degree of exertion * Pain may occur at rest * Rupture of unstable atherosclerotic plaque with subsequent platelet aggregation and thrombosis (partially occlusive) * Without myocardial necrosis * Can be a a precursor to myocardial infarction |
| variant angina | Variant angina (Prinzmetal angina) *May be no obvious plaques in arteries * Occlusion due to intense coronary artery spasm * Not exacerbated by physical exertion or emotion * Does not progress * Symptoms at rest, often occur at night Relatively rare |
| Stable Angina | Stable angina (angina pectoris) * Due to coronary artery plaque – usually with thick fibrous cap * When myocardial oxygen demand increases blood flow can not increase proportionally * Triggered by increase in oxygen requirements e.g. exercise * Short lasting pain 3 -15 minutes * No variation over last 2 months in: -frequency of symptoms -duration of symptoms - precipitating factors * Symptoms are consistent & predictable |
| outline classification of myocardial infarctions (STEMI, NSTEMI) | Myocardial infarction = “Heart attack” * Development of cardiac muscle necrosis resulting from an acute interruption of blood supply to a part of the heart **Symptoms of MI** No difference between STEMI and NSTEMI Similar to those for angina, but more severe & prolonged Chest pain, Tachycardia, Breathing difficulty, Nausea & vomiting, Sweating (diaphoresis) (STEMI) * Usually indicates complete occlusion & full-thickness heart muscle injury * Non-ST segment elevation myocardial infarction (NSTEMI) * Usually involves partial occlusion & partial thickness damage to heart muscle ** Considered a less severe type of MI |
| Describe triggers for angina (factors that increase myocardial oxygen requirements) | Stress/ emotion exercise |
| Recognise the symptoms of angina and myocardial infarction and understand how they develop with regard to the pathophysiology | Chest pain, Tachycardia, Breathing difficulty, Nausea & vomiting, Sweating |
| Describe the major classes of medications used in the management of IHD: | Antihypertensives (beta-blockers, calcium channel blockers), antiplatelets, anticoagulants & fibrinolytic drugs |
| antihypertensives | (beta-blockers, calcium channel blockers) |
| MOA of beta-blockers | Mode of action Competitively block beta receptors in heart, peripheral vasculature, bronchi, pancreas, uterus, kidney, brain and liver. Beta-blockers reduce heart rate, BP and cardiac contractility; also depress sinus node rate and slow conduction through the atrioventricular (AV) node, and prolong atrial refractory periods. The affinity of individual beta-blockers for beta receptors varies AEs Cardiac (e.g. bradycardia, hypotension) Off-target (e.g. bronchospasm) Other: insomnia, nightmares, depression Activation of beta-2 receptors causes vasodilation Activation of alpha-2 receptors causes vasoconstriction If block beta receptors, while alpha receptors remain unopposed → vessels susceptible to constriction |
| Beta Blockers Drug Interactions | Drug interactions Additive effects with other drugs that slow heart rate Digoxin, some calcium channel blockers Beta agonists (e.g. salbutamol used for asthma) Directly oppose beta-blocker effects Issue primarily with non-selective beta-blockers Anti-diabetic medications Beta receptors involved in response to hypoglycaemia |
| MOA of calcium channel blockers | Mode of action Block inward current of calcium into cells in vascular smooth muscle, myocardium and cardiac conducting system via L‑type calcium channels. Act on coronary arteriolar smooth muscle to reduce vascular resistance and myocardial oxygen requirements, relieving angina symptoms. Dihydropyridines act mainly on arteriolar smooth muscle to reduce peripheral vascular resistance and BP. They have minimal effect on myocardial cells. Non-dihydropyridines: diltiazem and verapamil act on cardiac and arteriolar smooth muscle. They reduce cardiac contractility, heart rate and conduction, with verapamil having the greater effect. Diltiazem has a greater effect on arteriolar smooth muscle than verapamil. |
| AEs Calcium Channel Blockers | Adverse effects: Depend on type/properties of CCB Headache, flushing, peripheral oedema Constipation, dyspepsia Gingival hyperplasia Overgrowth of gums Rare, most common with nifedipine Reversible when CCB ceased Most CCBs susceptible to CYP3A4 mediated drug interactions |
| explain the mechanism of action, common adverse effects and interactions of: nitrates | Mode of action Provide exogenous source of nitric oxide (which mediates vasodilator effects). Predominantly venodilators; reduce venous return and preload to the heart, reducing myocardial oxygen requirement. Indications Prevention and treatment of angina Chronic heart failure (isosorbide dinitrate with hydralazine) Acute heart failure associated with MI and unstable angina (glyceryl trinitrate infusion) Common adverse effects relate to vasodilation: Headache Flushing Dizziness Orthostatic hypotension Reflex tachycardia |
| explain the mechanism of action, common adverse effects and interactions of nicorandil | class: other antianginal drugs Mode of action Produces venous and arterial dilation due to its nitrate moiety and its effect on potassium channels in vascular smooth muscle; improves myocardial oxygen balance and decreases angina. Indications Prevention and treatment of stable angina |
| explain the mechanism of action, common adverse effects and interactions of ivabradine | Mode of action Ivabradine inhibits a current regulating the interval between depolarisations of the sinoatrial (SA) node. It reduces heart rate (by about 10 beats/minute) which in turn lowers cardiac workload and myocardial oxygen demand. Blocks sodium channels in the sino-atrial node Increases interval between depolarisation class: other abtianginal drugs From Lecture: Reduces heart rate Decreases myocardial oxygen demand Adverse effects Heart (predictable) = e.g. bradycardia, arrythmias Eye - luminous effects Metabolised by CYP450 = susceptible to drug interactions |
| explain the mechanism of action, common adverse effects and interactions of perhexiline | AMH: Mode of action Unclear. It inhibits carnitine palmitoyltransferase, which may change myocyte metabolism from long-chain fatty acids to glucose, leading to improved myocardial oxygen utilisation and anti-ischaemic effects. From lecture: Inhibits enzyme involved in long-chain fatty acid metabolism in the heart → Glucose metabolism predominates Oxidation of glucose requires less oxygen than oxidation of fatty acids = reduces myocardial oxygen requirements RISKS: Narrow therapeutic index Requires therapeutic drug monitoring Non-linear pharmacokinetics Adverse effects: neurotoxicity, hepatotoxicity Metabolised by polymorphic CYP2D6 = large interindividual variation in dose required |
| Briefly describe the surgical procedures used in the management myocardial infarction | . |
| Goals of treatment of ischaemic heart disease | Manage acute angina episodes safely & effectively Restore the balance between myocardial oxygen supply & demand Two strategies: Reduce oxygen demand of heart Increase blood flow by dilating coronary arteries (variant) Reduce frequency of angina episodes Delay progression of atherosclerosis/ischaemic heart disease Prevent myocardial infarction |
| Management of atherosclerosis & ischaemic heart disease | Dietary interventions Modification of risk factors Drugs to treat dyslipidaemia Beta-blockers and calcium channel blockers (antihypertensives) Drugs to manage angina: Nitrates, nicorandil, ivabradine, perhexiline Antithrombotic drugs (antiplatelets, anticoagulants) Fibrinolytic agents Angioplasty & other surgical interventions |
{kind=link}
Möchten Sie mit GoConqr kostenlos Ihre eigenen Karteikarten erstellen? Mehr erfahren.