366148
Description
Flashcards by Jon Ander Gil, updated more than 1 year ago
|
|
Created by Jon Ander Gil
about 11 years ago
|
|
| Question | Answer |
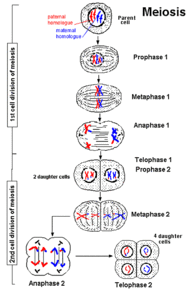
| MEIOSIS: Es una división celular en la que una célula diploide (2n) experimenta dos divisiones sucesivas, primera y segunda división meiótica (meiosis I y meiosis II), de las que se generan cuatro células haploides (n). Ambas divisiones comprenden profase, metafase, anafase y telofase. Se realiza en las glándulas sexuales para la producción de gametos. |
Image:
meiosis.gif (image/gif)
|
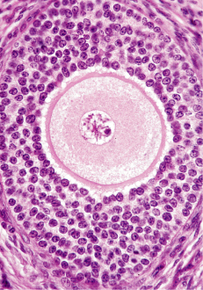
| OVOCITO PRIMARIO: Es una célula germinal femenina que está en proceso de convertirse en un óvulo maduro. Los ovocitos primarios se forman tras la ovocitogénesis antes del nacimiento. |
Image:
ROSS-PL-88d (image/jpg)
|
| ESPERMATOCITO PRIMARIO: Es una célula germinal masculina. Se forma en los testículos mediante la espermatocitogénesis a partir de las espermatogonias (células diploides (2n)). | |
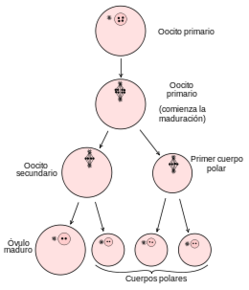
| CUERPOS POLARES: Son células que se forman por ovogénesis y sirven para la alimentación del embrión en la primera etapa del embarazo; no son funcionales y no pueden ser fecundados. La ovogonia se divide y forma un ovocito primario que, tras la meiosis, da lugar a un ovocito secundario y un cuerpo polar; tras una segunda meiosis II se forman el óvulo y tres cuerpos polares. |
Image:
Cuerpos_polares (image/png)
|
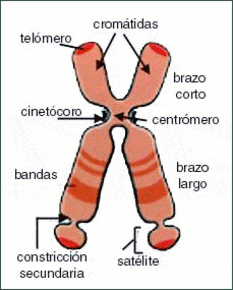
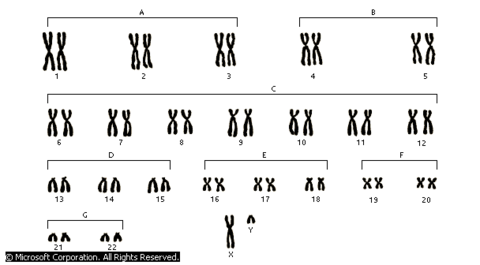
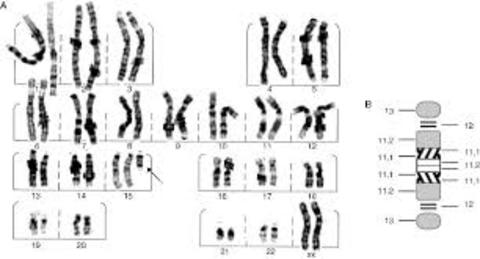
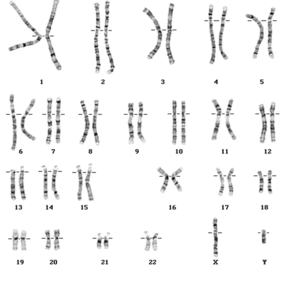
| CROMOSOMAS: Son cada uno de los pequeños cuerpos en forma de bastoncillos en que se organiza la cromatina del núcleo celular durante las divisiones celulares (mitosis y meiosis). Tiene una región condensada llamada centrómero, que confiere la apariencia general de cada cromosoma y que permite clasificarlos según la posición del centrómero a lo largo del cromosoma. El número de cromosomas de los individuos de la misma especie es constante. Cada cromosoma con una longitud y una posición del centrómero determinada existe otro cromosoma con rasgos idénticos: cromosomas homólogos. | |
| ANOMALÍAS CROMOSÓMICAS: Son defectos genéticos que generalmente se producen por desordenes en los cromosomas (estructura o cantidad). Pueden producir la muerte del niño antes de que nazca. Aproximadamente uno de cada 200 bebés nace con una anomalía cromosómica y pueden presentar problemas de conducta, retraso mental, incapacidades de aprendizaje, etc. | |
| DEFECTOS CONGÉNITOS: Son problemas que ocurren mientras un bebé se desarrolla dentro del cuerpo de su madre. La mayoría ocurren durante los primeros 3 meses del embarazo. Puede afectar al aspecto del cuerpo, a su funcionamiento o ambos. Pueden ser causados por la exposición a medicinas, sustancias químicas o infecciones durante el embarazo. El ácido fólico puede prevenir algunos defectos congénitos. Actualmente, se pueden diagnosticar muchos defectos congénitos durante el embarazo, lo que permite tratar y corregir algunos problemas antes del nacimiento. | |
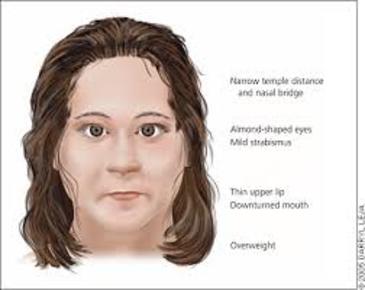
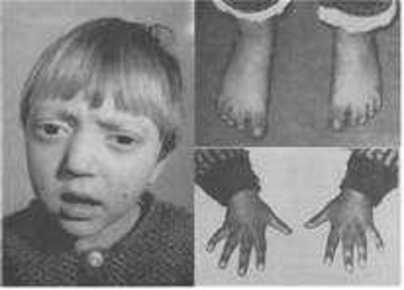
| SÍNDROME DE TURNER: Es una enfermedad genética caracterizada por la presencia de un solo cromosoma X. Es la única monosomía viable en humanos. La ausencia de cromosoma Y determina el sexo femenino de los individuos afectados, y la ausencia del segundo cromosoma X determina la falta de desarrollo de los caracteres sexuales primarios y secundarios; esto confiere un aspecto infantil e infertilidad de por vida. | |
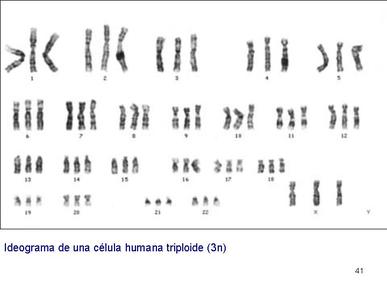
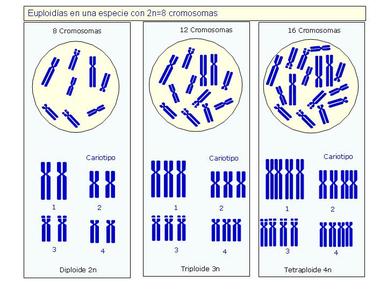
| TRIPLOIDÍA: Es la presencia de una dotación cromosómica de 3n cromosomas, frente a los 2n normales de las células diploides. Se produce por la falta de disyunción en la formación de uno de los gametos de los padres, de modo que tendrá carga doble en el gameto que aporte. |
Image:
Triploid_a.JPG (image/JPG)
|
| TRISOMÍA: Es la existencia de un cromosoma extra en un organismo diploide, de modo que en vez de un par homólogo de cromosomas es un triplete (2n + 1 cromosomas). La adición de un cromosoma extra X o Y a una mujer o un varón da lugar a individuos viables que presentan diversos síndromes (el síndrome de Klinefelter, el síndrome del triple X o el síndrome del XYY). La adición de un autosoma a la dotación diploide tiene graves efectos y normalmente es letal durante su desarrollo. |
Image:
Trisom_a (image/jpg)
|
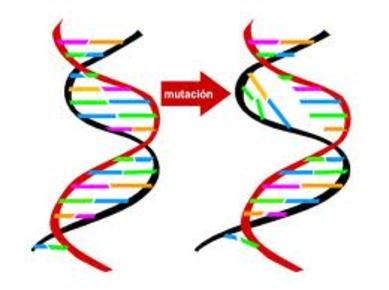
| MUTACIÓN DE GENES: Es una alteración o cambio en la información genética (genotipo) de un ser vivo (muchas veces por contacto con mutágenos) que va a producir un cambio de características de éste; se presenta súbita y espontáneamente, y se puede transmitir a la descendencia. | |
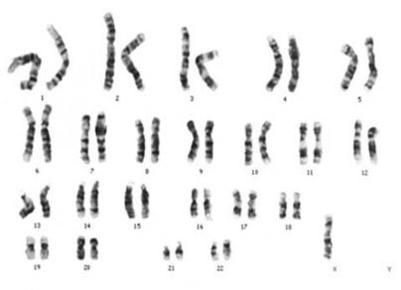
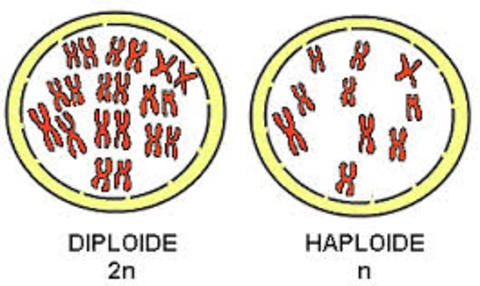
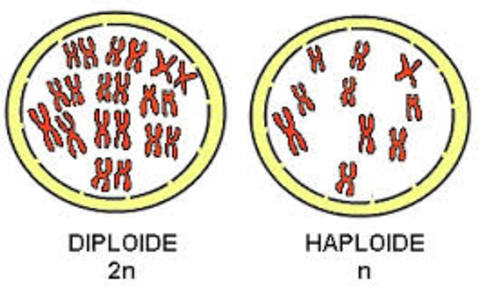
| DIPLOIDE: Las células diploides (2n) son las células que tienen un número doble de cromosomas Las células somáticas del ser humano contienen 46 (23 x 2) cromosomas, siendo éste su número diploide. |
Image:
Diploide (image/jpg)
|
| HAPLOIDE: Las células haploides son las células que contiene un solo juego de cromosomas (n). Los gametos tienen solamente la mitad, 23, lo que constituye su número haploide. |
Image:
Diploide (image/jpg)
|
| EUPLOIDÍA: Es una alteración numérica en la dotación total de cromosomas, de manera que los cromosomas de más o de menos constituyen un juego completo. |
Image:
Euploid_a.JPG (image/JPG)
|
| ANEUPLOIDÍA: Es una alteración numérica en la dotación total de cromosomas, de manera que los cromosomas de más o de menos no corresponden a un juego completo. Una de las aneuploidías más comunes es el síndrome de Down, que es una trisomía del cromosoma 21; también se puede observar frecuentemente en células cancerosas. |
Image:
Aneuploid_a (image/jpg)
|
| MONOSOMÍA: Es una anomalía cromosómica caracterizada por la pérdida de un cromosoma, lo que da lugar a una dotación cromosómica (2n – 1). En la especie humana los individuos con tales dotaciones cromosómicas no sobreviven al desarrollo embrionario y fetal, pero sí hay casos de supervivientes con monosomías parciales. |
Image:
Monosom_a (image/png)
|
| DISYUNCIÓN: Es la separación de los cromosomas homólogos apareados durante el estado de anafase de la primera división meiótica, o de las cromátidas durante la anafase de la mitosis y la segunda división meiótica. |
Image:
Disyunci_n (image/png)
|
| MOSAICISMO: Es una alteración genética, resultado de un error en la división celular muy temprano durante desarrollo fetal, en la que, en un mismo individuo, coexisten dos o más poblaciones de células con distinto genotipo, supuestamente originadas a partir de un mismo cigoto. Esta situación puede afectar a cualquier tipo de célula. |
Image:
Mosaicismo (image/jpg)
|
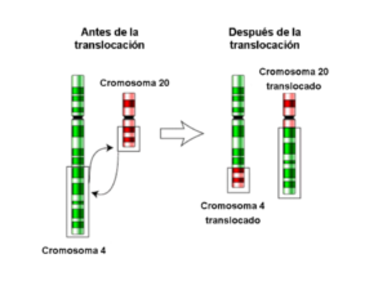
| TRANSLOCACIÓN: Es el cambio de posición de un fragmento de cromosoma a otro cromosoma no homólogo del mismo núcleo o a otra parte del mismo. El modo más fácil para que ocurra esto es que los brazos de dos cromosomas no homólogos se aproximen de tal manera que se facilite el intercambio. No hay perdida o ganancia de información genética; solo hay una reordenación del material genético. La presencia de una traslocación no afecta directamente a la viabilidad de los individuos que la llevan. |
Image:
Translocaci_n (image/png)
|
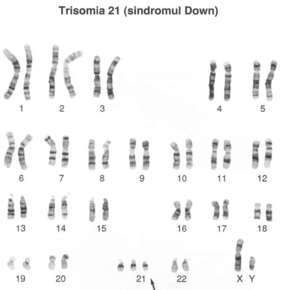
| TRISOMÍA 21 (SÍNDROME DE DOWN): Es la trisomía en la que hay una copia extra del cromosoma 21 el cual causa problemas en el desarrollo del cuerpo y cerebro. El síndrome de Down es una de las causas más comunes de anomalías congénitas en los humanos y es la única trisomía autosómica de la especie humana de la que sobrevive un número significativo de individuos más allá del año después del nacimiento. La incidencia global del síndrome de Down se aproxima a uno de cada 700 nacimientos, pero el riesgo aumenta con la edad de la madre. |
Image:
Trisom_a_21 (image/png)
|
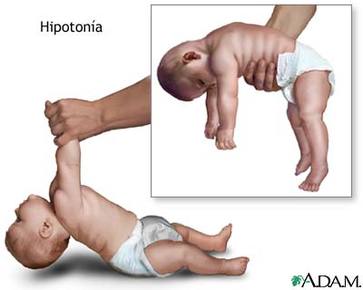
| HIPOTONÍA: Es la disminución del tono muscular en forma generalizada o focal, que generalmente se asocia a déficit en el desarrollo psicomotor. Este síndrome se caracteriza por la presencia de posturas anormales y poco habituales, disminución de la resistencia de las articulaciones a los movimientos pasivos, aumento de la movilidad de las articulaciones, o amplitud durante los movimientos pasivos. |
Image:
Hipoton_a (image/jpg)
|
| LEUCEMIA: Es la proliferación incontrolada de las células sanguíneas inmaduras en la médula ósea, acumulándose en ésta y en la sangre, de modo que reemplazan a las células normales. Por tratarse de una proliferación de células inmaduras y anormales en la sangre se le considera un "cáncer de la sangre". Es el cáncer más frecuente en la infancia, con 3-5 casos por año por cada 100.000 niños menores de 15 años. Las causas se desconoce en la mayoría de los casos pero está demostrado que no es un padecimiento hereditario o contagioso; la mayor parte de las veces se presenta en niños previamente sanos. |
Image:
leucemia (image/jpg)
|
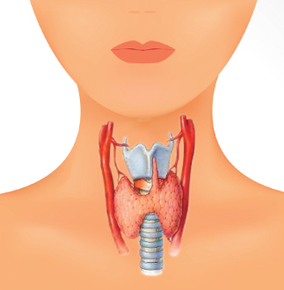
| DISFUNCIÓN TIROIDEA: Es el mal funcionamiento de la glándula tiroides la cual, mediante la producción de las hormonas tiroxina y triyodotironina, controla el metabolismo. Si disminuye su actividad, disminuye la producción de hormona y el metabolismo se enlentece; si aumenta su actividad, aumenta la producción de hormona y el metabolismo se acelera. | |
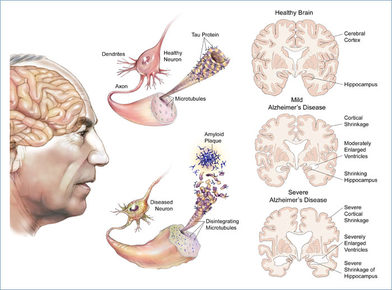
| ALZHEIMER: Es una enfermedad neurodegenerativa que se manifiesta como deterioro cognitivo y trastornos conductuales, producidos por la pérdida gradual de neuronas cerebrales y la atrofia de diferentes zonas del cerebro. Se caracteriza, en su forma típica, por una pérdida de la memoria inmediata y de otras capacidades mentales dado que afecta a las partes del cerebro que controlan el pensamiento, la memoria y el lenguaje. Suele tener una duración media aproximada, después del diagnóstico, de 10 años. Es la forma más común de demencia, es incurable y terminal, se desconoce la causa exacta de la misma y aparece con mayor frecuencia en personas mayores de 65 años. | |
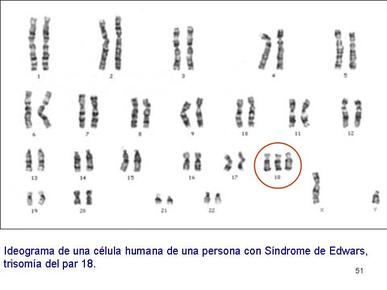
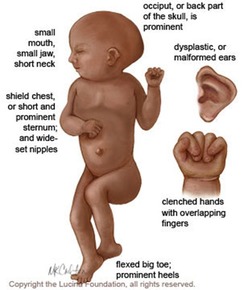
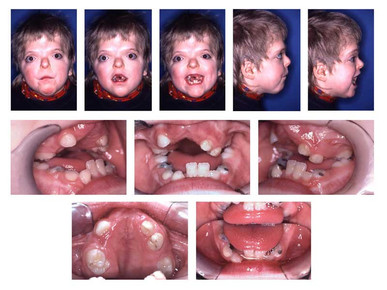
| TRISOMÍA 18 (SÍNDROME DE EDWARDS): Es un trastorno genético en el cual una persona tiene una tercera copia del material del cromosoma 18, en lugar de las dos copias normal, lo cual afecta el desarrollo normal. Es bastante común y tres veces más frecuente en niñas que en niños. Dada la alta tasa de mortalidad postnatal de esta enfermedad genética, no existe a día de hoy un tratamiento eficaz. El riesgo de concebir un niño con síndrome de Edwards incrementa a medida que lo hace la edad de la madre. |
Image:
Trisom_a_18 (image/jpg)
|
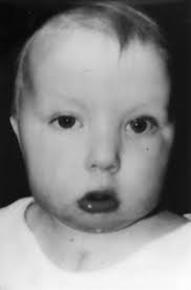
| MICROGNATIA: Es una alteración del desarrollo de la mandíbula fetal caracterizado por un tamaño menor de lo normal. Habitualmente se asocia con retrognatia (posición anormalmente retrasada de la mandíbula respecto al maxilar superior). Puede responder a una causa mandibular primaria, o asociarse a enfermedades esqueléticas o neuromusculares que produzcan contractura fija de la articulación temporomaxilar, a alteraciones cromosómicas (fundamentalmente trisomía 18 y triploidías) o a una gran variedad de síndromes. De todas ellas, las alteraciones cromosómicas son la causa más frecuente. |
Image:
Micrognatia (image/jpg)
|
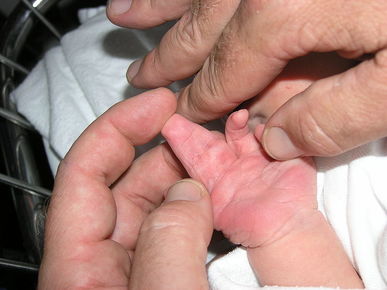
| SINDACTILIA: Es la fusión de dos o más dedos de las manos o de los pies; generalmente implica la conexión cutánea entre las dos áreas, pero en casos muy poco frecuentes implica la de los huesos. |
Image:
Sindactilia.JPG (image/JPG)
|
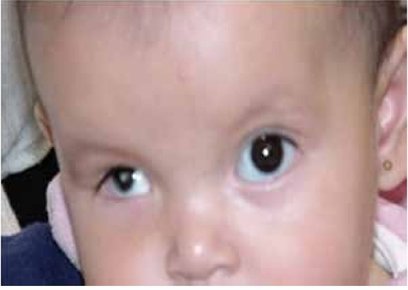
| MICROFTALMÍA: Es una anomalía ocular en la que uno o ambos globos oculares tienen un tamaño muy pequeño. En algunos afectados el globo ocular parece inexistente, pero, aún en estos casos, existe algún tejido ocular. Puede provocar o no la pérdida de visión, según el grado de afectación del globo ocular. Entre un tercio y la mitad de los individuos afectados de microftalmía la tienen como parte de un síndrome que afecta a otros órganos y tejidos corporales. Puede estar causada por mutaciones en muchos genes implicados en el desarrollo precoz del ojo, la mayoría de los cuales no han sido identificados. |
Image:
Microftalm_a (image/jpg)
|
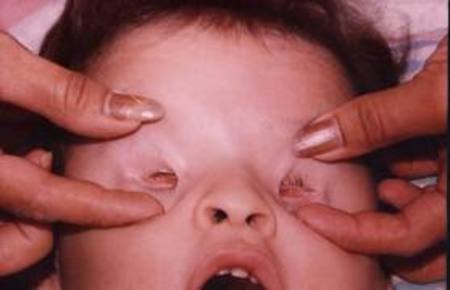
| ANOFTALMÍA: O ausencia completa del globo ocular, es una malformación a nivel orbitario ya sea de forma congénita o adquirida, que conlleva a la perdida de la función ocular y visión. Está relacionada con causas ambientales, virus, fiebre, abuso de disolventes, exposición a radiación o drogas y con síndromes genéticos. |
Image:
Anoftalm_a (image/jpg)
|
| COLOBOMA: Es la falta parcial del tejido normal del ojo en el momento del nacimiento El coloboma puede afectar uno o ambos ojos. Si la afección está presente en ambos ojos, el coloboma puede afectarlos de igual o diferente forma. Dependiendo de cuál es la parte afectada del ojo, existen varios tipos: Puede ser hereditaria o aparecer sin historia familiar previa. |
Image:
coloboma (image/jpg)
|
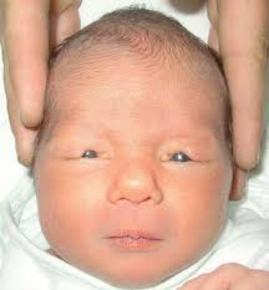
| OCCIPUCIO: Es la parte posterior de la cabeza por la que ésta se une con las vértebras del cuello. Un occipucio prominente es característico en la Trisomía 18 y en la Trisomía 9. |
Image:
Occipucio (image/jpg)
|
| LABIO LEPORINO: Es un defecto congénito que consiste en una hendidura o separación en el labio superior, originado por fusión incompleta de los procesos maxilar y nasomedial del embrión y es uno de los defectos de nacimiento más frecuentes. Se presenta, frecuentemente, acompañado de paladar hendido. |
Image:
Labio_leporino (image/jpg)
|
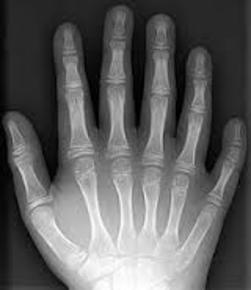
| POLIDACTILIA: Es un trastorno genético por el que el individuo nace con más dedos en la mano o en el pie de los que le corresponde (por lo general un dedo más). Puede ocurrir espontáneamente, sin ningún otro síntoma o enfermedad. Se puede transmitir de padres a hijos. |
Image:
polidactilia (image/jpg)
|
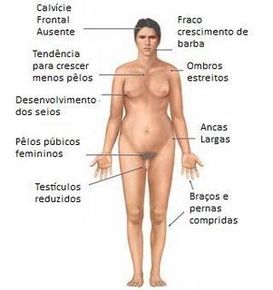
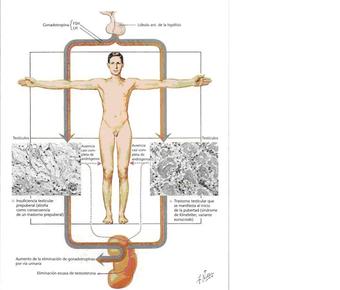
| SÍNDROME DE KNIFELTER: Es una anomalía cromosómica que afecta solamente a los hombres los cuales presentan un cromosoma X adicional en la mayoría de sus células. Puede afectar a las diversas etapas del desarrollo físico, social y del lenguaje. El síntoma más común es la infertilidad. Generalmente no producen la misma cantidad de testosterona lo que ocasiona menos vello facial y corporal y ser menos musculosos. | |
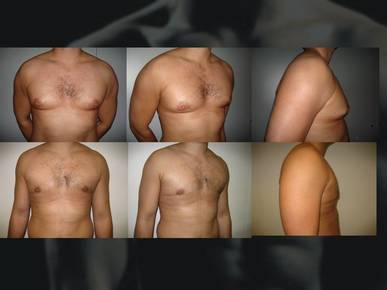
| GINECOMASTIA: Es el engrandecimiento patológico de una o ambas glándulas mamarias en el hombre debido al crecimiento excesivo del tejido mamario y no al exceso de tejido adiposo. Este trastorno suele estar asociado a una hiperprolactinemia o a un hiperestrogenismo .Se puede dar en una o en ambas mamas. La ginecomastia durante la pubertad es común y generalmente desaparece en un período de meses. | |
| DISGENESIA GONADAL: Desarrollo anormal de la gónada fetal. Se suele asociar a alteraciones del desarrollo de los gonaductos internos y de los genitales externos, lo que conduce, en ocasiones, a estados de ambigüedad sexual. | |
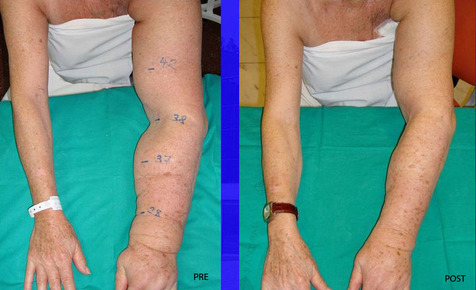
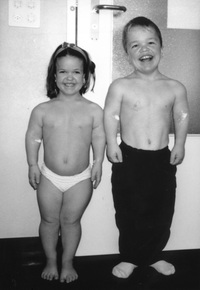
| LINFEDEMA: Es un tipo de edema producido por una obstrucción en los canales linfáticos del organismo como consecuencia de la acumulación de la linfa (rica en proteínas y fibroblastos) en los espacios intersticiales del tejido celular subcutáneo. Generalmente es debido a un fallo o a una insuficiencia en el sistema linfático, y trae como consecuencia el aumento del volumen de las extremidades. |
Image:
linfedema_B (image/jpg)
|
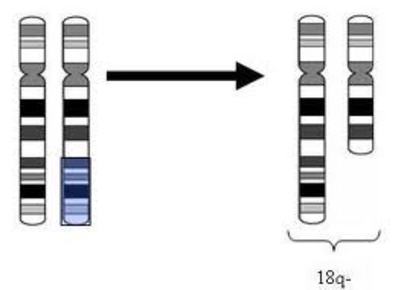
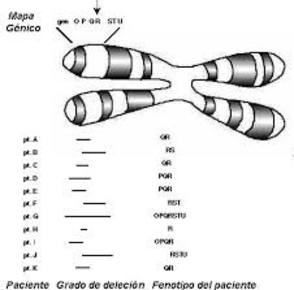
| DELECIÓN: Es un tipo especial de anomalía estructural cromosómica que consiste en la pérdida de un fragmento de ADN en el extremo o a lo largo de uno de los brazos de un cromosoma, lo que origina un desequilibrio. |
Image:
Deleci_n (image/jpg)
|
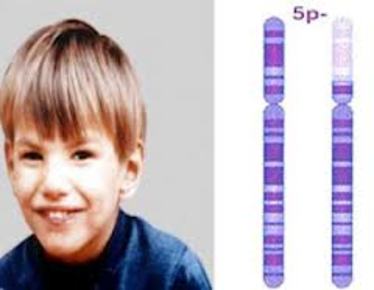
| SÍNDROME DE MAULLIDO DE GATO: (o síndrome de Lejeune),es una enfermedad congénita infrecuente con alteración cromosómica provocada por un tipo de deleción autosómica terminal o intersticial del brazo corto del cromosoma 5, caracterizada por un llanto que se asemeja al maullido de un gato y que se va modificando con el tiempo. Predomina en las niñas. | |
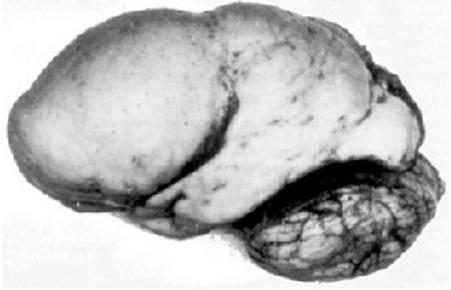
| MICROCEFALIA: Es una afección en la cual la cabeza de una persona es considerablemente más pequeña de lo normal para su edad y sexo. Se presenta cuando el cerebro no logra crecer a una tasa normal, ya que el crecimiento del cráneo está determinado por el del cerebro, el cual tiene lugar en el útero y durante la lactancia. Las enfermedades que afectan el crecimiento cerebral (infecciones, trastornos genéticos y desnutrición grave) pueden ocasionar microcefalia. |
Image:
Microcephaly (image/png)
|
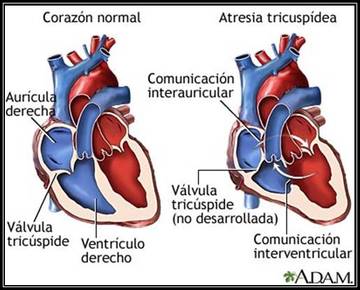
| CARDIOPATÍA: Es la enfermedad propia de las estructuras del corazón. Existen factores de riesgo que aumentan la probabilidad de contraer esta enfermedad que no son controlables (edad, sexo, genes o raza) y otros que sí lo son (tabaco, colesterol, hipertensión, peso, ejercicio, alimentación sana, alcohol, estrés). | |
| SÍNDROME DE MICRODELECIONES O SÍNDROME DE LOS GENES CONTIGUOS: Es el síndrome causado por una deleción que incluye varios genes distintos en un mismo segmento cromosómico. A menudo se presenta como una microdelección. | |
| BANDEO DE CROMOSOMAS DE ALTA RESOLUCIÓN: Los estudios de bandeo extendido o de "alta resolución" implican el estudio de los cromosomas con una resolución más alta que la del análisis cromosómico estándar. Los cromosomas están dispuestos de tal manera que se alargan un poco, por lo que se pueden ver más bandas. Esto permite observar partes más reducidas del cromosoma e identificar, de este modo, anomalías cromosómicas estructurales más pequeñas que no pueden ser vistas en un análisis de rutina. | |
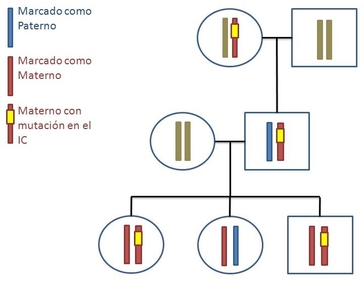
| SÍNDROME DE PRADER-WILLI: Es una enfermedad congénita causada por la carencia de un gen del cromosoma 15. Normalmente, cada uno de los padres transmite una copia de este cromosoma; la mayoría de los pacientes con este síndrome carecen del material genético en parte del cromosoma del padre; el resto de los pacientes con frecuencia tiene dos copias del cromosoma 15 de la madre. Los cambios genéticos ocurren en forma aleatoria, sin antecedentes familiares. Son personas obesas, con disminución del tono muscular y de la capacidad mental y con glándulas sexuales que producen pocas o ninguna hormona. | |
| HIPOGONADISMO: Es un trastorno en el que los testículos u ovarios no son funcionales o hay incapacidad genética del hipotálamo para secretar cantidades normales de hormona liberadora de gonadotropina. Las características sexuales masculinas o femeninas no están desarrolladas: huesos finos, músculos débiles, vello leve, la voz se reduce un poco a su tono grave. Es causas de esterilidad. | |
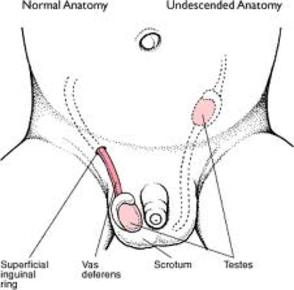
| CRIPTORQUIDIA: Es un trastorno del desarrollo que consiste en el descenso incompleto de uno o ambos testículos a través del canal inguinal hacia el escroto. Si no se trata conduce a una esterilidad permanente y a una mayor probabilidad de desarrollar tumores testiculares. |
Image:
Criptorquidia (image/jpg)
|
| IMPRONTA GENÓMICA: Es un proceso biológico por el que un gen o dominio genómico se encuentra marcado bioquímicamente indicando su origen parental. | |
| SÍNDROME DE MILLER-DIEKER: Es una rara enfermedad congénita y hereditaria causado por una mutación en el brazo corto del cromosoma 17. Los afectados presentan problemas en el desarrollo del sistema nervioso central, lo que conduce a alteraciones severas en la función neurológica. | |
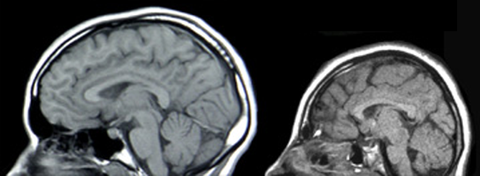
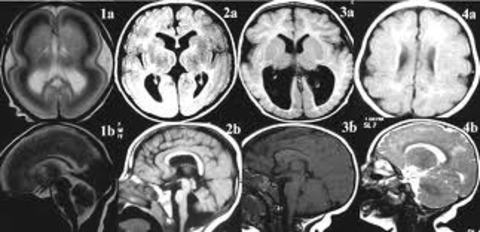
| LISENCEFALIA: Es un trastorno poco común de la formación del cerebro caracterizado por microcefalia y agiria (ausencia de las circunvoluciones normales del cerebro). Es causada por una migración neuronal defectuosa, de modo que las células nerviosas se desplazan desde el lugar de origen a su localización permanente. Puede ser debido a infecciones virales, a la escasez de riego sanguíneo al cerebro del feto o a un trastorno genético. |
Image:
Lissencephaly (image/jpg)
|
| SÍNDROME VELOCARDIOFACIAL (O SÍNDROME DE DIGEORGE): Es una enfermedad causada por la deleción de una pequeña parte del cromosoma 22. Da lugar a diferentes cuadros según el individuo que lo presente pero son comunes a todos ellos los defectos cardiacos, el aspecto del rostro y ausencia o subdesarrollo del timo y las glándulas paratiroides. | |
| TRISOMÍA 16: (47 + XY), es la trisomía más frecuente, ya que se da en el 1% de las concepciones, pero totalmente inviable, dando lugar a un aborto alrededor del tercer mes. | NO SE HA ENCONTRADO IMÁGEN |
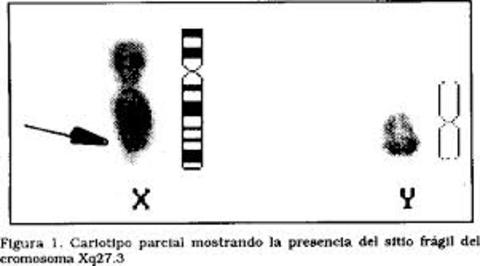
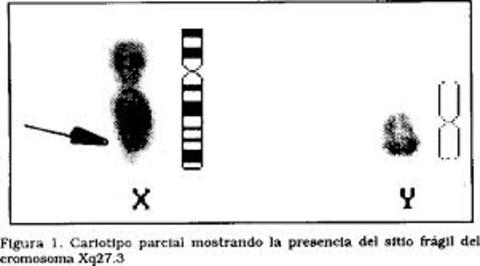
| SITIOS FRÁGILES EN LOS CROMOSOMAS: Son unas regiones específicas de los cromosomas que tienen tendencia a romperse. Constituyen áreas de cromatina que no se compactan apropiadamente durante la mitosis debido a que presentan una replicación más tardía que el resto del cromosoma. Su ubicación es la misma en todas las células de un individuo o de una familia. | |
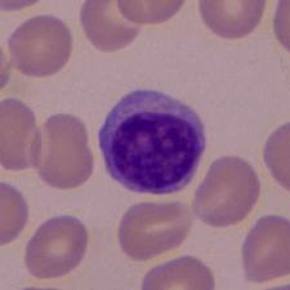
| LINFOCITOS: Es una célula linfática (se fabrican por células linfoides presentes en la médula ósea ), que es un tipo de leucocito (glóbulo blanco) agranulocito. Son los leucocitos de menor tamaño y representan aproximadamente el 30% del total en la sangre periférica. Presentan un gran núcleo esférico y escaso citoplasma que contiene algunas mitocondrias, ribosomas libres y un pequeño aparato de Golgi. Son células circulantes del sistema inmunitario que reaccionan frente a materiales extraños y son encargadas de la inmunidad específica o adquirida. Se localizan fundamentalmente en la linfa y los órganos linfoides y en la sangre. |
Image:
linfocitos (image/jpg)
|
| SÍNDROME DE X FRÁGIL: (o síndrome de Martin-Bell), es un trastorno hereditario que ocasiona retraso mental, debido a una mutación conocida como expansión de repeticiones de trinucleótidos, que supone el incremento en la descendencia del número de repeticiones de tres bases del ADN. La mutación que origina el síndrome afecta a una región del cromosoma X en la que se sitúa el gen FMR-1 (gen que produce una proteína necesaria para que el cerebro crezca apropiadamente; si el gen es defectuoso, no se produce proteína). Cuando el número de repeticiones supera el valor umbral de 230 repeticiones se produce la metilación del gen y, por tanto, éste pierde su función, produciendo así el síndrome del X frágil. | |
| ALELOS: Es cada una de las formas alternativas que puede tener un gen, que se diferencian en su secuencia y que se puede manifestar en modificaciones concretas de la función de ese gen. Al ser la mayoría de los mamíferos diploides estos poseen dos cromosomas, uno de ellos procedente del padre y el otro de la madre. Cada par de alelos se ubica en igual locus o lugar del cromosoma. |
Image:
alelos (image/jpg)
|
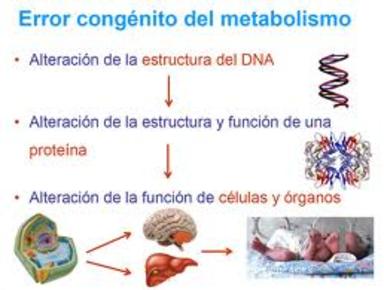
| ERRORES CONGÉNITOS DEL METABOLISMO: Son un conjunto de enfermedades hereditarias que implican alteraciones del metabolismo. La mayoría son debidas a la alteración de un gen que codifica un enzima que cataliza una de los miles de reacciones químicas de la célula. | |
| FENILCETONURIA: Es un error congénito del metabolismo causado por la carencia de la enzima fenilalanina hidroxilasa, lo que se traduce en la incapacidad de metabolizar el aminoácido tirosina a partir de fenilalanina en el hígado; la fenilalanina se acumula y resulta tóxica para el sistema nervioso central, ocasionando daño cerebral. Es una enfermedad congénita con un patrón de herencia recesivo. Es un tipo de hiperfenilalaninemia. | |
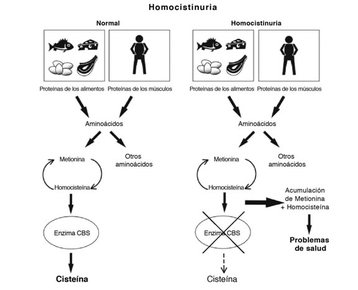
| HOMOCISTINURIA: Es un grupo de enfermedades metabólicas poco frecuentes que se caracterizan por presentar un nivel elevado del aminoácido homocisteína en el plasma sanguíneo, lo que conlleva un aumento de homocisteína en la orina. Los niveles de homocisteína elevados en sangre guardan una estrecha relación con ateroesclerosis prematura, trombosis recurrentes de arterias coronarias, cerebrales o periféricas, trombosis venosa y con el riesgo de desarrollar mal de Alzheimer. |
Image:
Homocistinuria (image/jpg)
|
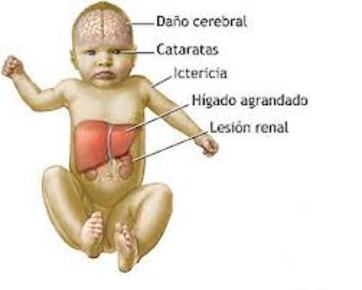
| GALACTOSEMIA: Es una enfermedad hereditaria causada por una deficiencia enzimática y se manifiesta con incapacidad de utilizar el azúcar galactosa, lo cual provoca una acumulación de éste en el organismo, produciendo lesiones en el hígado y sistema nervioso central. |
Image:
galactosemia (image/jpg)
|
| BANDEO DE GIEMSA: Es una técnica para la tinción de cromosomas basada en el tratamiento con tripsina y posterior tinción con el colorante Giemsa. Los cromosomas teñidos presentan una secuencia nítida de bandas claras y oscuras con la ventaja de ser permanente y observable al microscopio de luz, lo que permite hacer un estudio de los cromosomas. |
Image:
Bandeo_de_giemsa (image/png)
|
| SÍNDROME DE WAARDENBURG: Es una enfermedad rara hereditaria, con carácter autosómico dominante que se caracteriza por anomalías faciales, oculares y sordera neurosensorial. | |
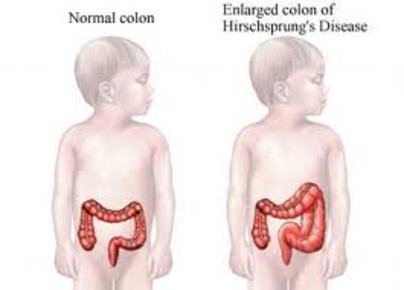
| ENFERMEDAD DE HIRSCHSPRUNG: Es un trastorno congénito que produce una obstrucción del intestino grueso debido al movimiento muscular impropio en el intestino. Se da una ausencia de parte de los nervios de las capas musculares del intestino por lo que en estas zonas, al no producirse contracciones musculares, los materiales digeridos no pueden ser movilizados causando un bloqueo; los contenidos intestinales se acumulan detrás del bloqueo, haciendo que el intestino y el abdomen se hinchen. | |
| ENANISMO DE LARON: Es un desorden genético autosómico recesivo caracterizado por una insensibilidad a la hormona del crecimiento causada por una variación del receptor de la hormona. Esta condición causa baja estatura y resistencia al cáncer. | |
| SÍNDROME MANO-PIE-GENITAL: Es una enfermedad causada por una mutación autosómica dominante que se caracteriza por malformaciones en las extremidades y defectos urogenitales. | NO SE HA ENCONTRADO IMÁGEN |
| SÍNDROME DE PFEIFFER: Es una rara enfermedad de origen genético asociada a la fusión prematura de las suturas del cráneo (craneosinostosis) y a la fusión parcial de los dedos de los pies y de las manos (sindactilia). Se transmite de padres a hijos según un patrón autosómico dominante y puede estar causado por dos tipos de mutaciones, en el cromosoma 8 o en el cromosoma 10. | |
| SÍNDROME DE JACKSON WEIS: Es una rara enfermedad caracterizada por anomalías craneofaciales y una sindactilia parcial debida a una fusión simétrica entre algunos de los dedos de pies y manos. Se hereda como un rasgo genético autosómico dominante. | |
| SÍNDROME DE CROUZON: Es un trastorno genético que provoca la fusión anormal entre los huesos en el cráneo y rostro. Normalmente, a medida que el cerebro de un niño crece, las estructuras abiertas entre los huesos permiten que el cráneo se desarrolle normalmente. Cuando las estructuras se unen demasiado temprano, como en este caso, el cráneo crece en dirección de las estructuras abiertas restantes lo que provoca una cabeza, rostro, y dientes de forma anormal. Se transmite de padres a hijos según un patrón de herencia autosómico dominante. Está originado por una mutación de un gen que codifica el factor de crecimiento de los fibroblastos tipo 2 y 3. | |
| SÍNDROME DE APERT: Es un tipo de acrocefalosindactilia, un desorden congénito caracterizado por deformaciones en el cráneo, cara, manos y pies. Afecta al primer arco branquial que es precursor del maxilar y mandíbula. Se puede trasmitir de padres a hijos como un rasgo autosómico dominante . Es causado por mutaciones en el gen receptor 2 del factor de crecimiento de fibroblastos. Esta anomalía en los genes provoca que algunas de las suturas óseas del cráneo se cierren demasiado temprano. | |
| ACONDROPLASIA: Es un trastorno genético que causa enanismo y está causado por mutaciones en el gen FGFR3 el cual evita el crecimiento de cartílago en la placa de crecimiento. El FGFR3 codifica una proteína llamada receptor 3 del factor de crecimiento de fibroblastos la cual es el lugar de acción de un factor de crecimiento principal responsable del alargamiento de los huesos. Cuando este factor de crecimiento no puede actuar correctamente por la ausencia de su receptor, el crecimiento de los huesos se hace más lento lo que conlleva a huesos más cortos y en forma anormal, y estatura más corta. |
Image:
acondroplasia (image/jpg)
|
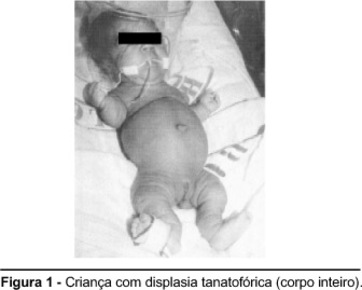
| DISPLASIA TANATOFÓRICA: Es la forma letal más común de displasia esquelética en el periodo neonatal caracterizada por un severo acortamiento de los miembros, tórax estrecho, macrocefalia y una longitud normal del tronco. La herencia es autosómica dominante, producida por una mutación a nivel del receptor 3 del factor de crecimiento de los fibroblastos. | |
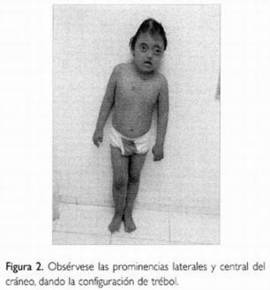
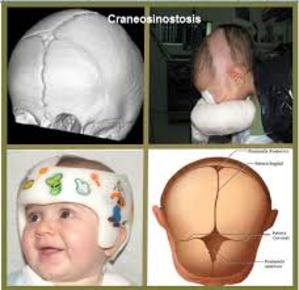
| CRANEOSINOSTOSIS: Es una alteración congénita en la que se produce el cierre prematuro de una o más de las suturas que separan los huesos del cráneo de un bebé. |
Image:
Craneosinostosis (image/jpg)
|
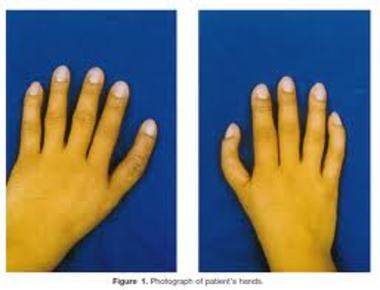
| SÍNDROME DE HOLT-ORAM: Es una enfermedad genética rara caracterizada por malformaciones distintivas de los huesos de los pulgares y de los antebrazos (miembros superiores) y por anomalías del corazón. Se debe a una mutación dominante en el gen TBX5 que altera la estructura tridimensional de la proteína impidiendo su correcta unión al ADN. | |
| SÍNDROME DE COLOBOMA RENAL: Es una enfermedad genética caracterizada por anomalías oculares y alteraciones renales. Se hereda mediante un patrón autosómico dominante. | NO SE HA ENCONTRADO IMAGEN CON AMBAS ALTERACIONES |
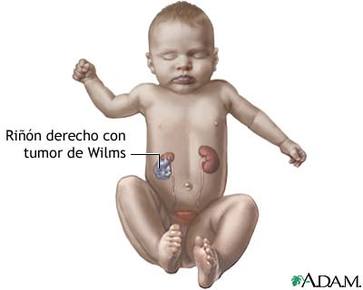
| TUMOR DE WILMS: Es un tipo de cáncer renal que se presenta en los niños menores de ocho años. Un 75% de los casos se presentan en niños sanos, mientras un 25% se asocian a anormalidades del desarrollo. Responde muy bien al tratamiento médico. |
Image:
Tumor_de_wilms (image/jpg)
|
| HIBRIDACIÓN IN SITU POR FLUORESCENCIA (FISH): Es una técnica molecular que permite localizar un determinado fragmento de la secuencia de los ácidos nucleicos y pone de manifiesto la presencia o ausencia de secuencias génicas específicas. Se puede aplicar sobre núcleos interfásicos de extensiones celulares, cortes de tejido o directamente en cromosomas. | |
| SÍNDROME DE SWYER: (o disgenesia gonadal XY) es una disgenesia gonadal pura, con cariotipo 46XY, fenotipo femenino normal y ausencia completa de tejido gonadal funcionante. Se caracteriza por una falta de correlación entre el fenotipo sexual manifestado – femenino- y el genotipo – XY, masculino-. En el nacimiento, los pacientes presentan un fenotipo femenino normal. Pero en la pubertad, normalmente no desarrollan, o lo hacen con retraso, características sexuales secundarias, tienen amenorrea y presentan un riesgo incrementado de padecer un tumor gonadal. |
Image:
swyer (image/png)
|
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Want to create your own Flashcards for free with GoConqr? Learn more.