29748866
Description
Flowchart by nallely gallegos, updated more than 1 year ago
|
|
Created by nallely gallegos
over 3 years ago
|
|
Flowchart nodes
- HERENCIA MENDELIANA
- HERENCIA LIGADA AL SEXO Aquellas enfermedades cuyos genes se localizan en los cromosomas sexuales X e Y
- INACTIVACIÓN DEL CROMOSOMA X Por mecanismos regulatorios genéticos y epigenéticos se induce el silencio transcripcional en uno de los cromosomas X en las células somáticas de las mujeres.
- Primera hipótesis por la Dra. Mary Lyon (1966). La inactivación sería fija y transmitida a todas las células hijas en cada ronda de división mitótica
- Corpúsculo de Barr: representa un cromosoma X muy condensado o inactivo, observados en estudios de citogenética en células en inferfase
- 2 formas de inactivación
- Centro de inactivación del cromosoma X (XIC)
- Localizado en Xq13.2, contiene un gen llamado XIST que se expresa sólo en el cromosoma inactivo y cuyo producto es un RNA no codificante encargado de silenciar el cromosoma que lo contiene.
- La inactivación será aleatoria del X paterno y del materno y su objetivo es minimizar las consecuencias clínicas de un defecto cromosomico
- No todos los genes son sujetos a inactivación.
- Inactivación de cromosomas sexuales en meiosis
- Silenciamiento transcripcional en los cromosomas sexuales del varón durante el paquiteno de la espermatogénesis.
- Inactiva a los cromosomas que fallan en el apareamiento con sus homólogos como protección de una subsecuente aneuploidía en generaciones celulares siguientes.
- Su falla se ha propuesto como mecanismo etiológico en la esterilidad meiótica del varón
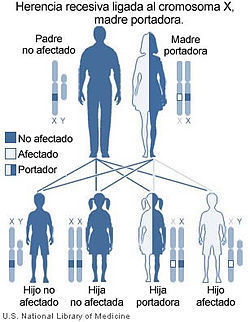
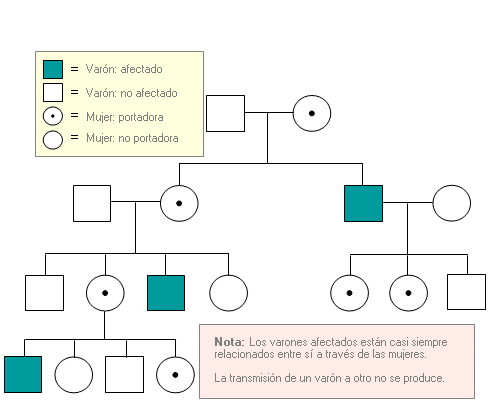
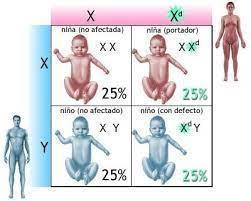
- HERENCIA RECESIVA LIGADA AL CROMOSOMA X
- Mujeres: Heredan dos copias del cromosoma X, pueden ser homocigotas o heterocigotas.
- Varones: Estará afectado por ser hemicigoto, aunque el gen sea recesivo, el cromosoma Y no proporciona la contraparte alélica normal
- Riesgos de recurrencia dependientes del genotipo de la pareja
- Varón afectado + mujer sana = todos hijos sanos
- Unión más frecuente: portadora con un varón sano
- Poco frecuente: Varón sano + portadora heterocigota
- Fenotipo anormal
- Árbol genealógico: destacan varones afectados emparentados por rama materna y saltos generacionales.
- Ejemplos destacables: Distrofia muscular de Duchenne, su variante alélica de Becker y la hemofilia
- Otros con menor frecuencia
- SÍNDROME DE LESCH-NYHAN (SLN)
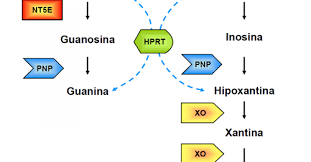
- Deficiencia completa o parcial de la actividad enzimática de la hipoxantina-guanina-fosforibosil-transferasa es un error innato del metabolismo de las purinas que no se reciclan y se degradan a ácido úrico, el cual presenta una sobreproducción por el incremento en la síntesis de purinas
- Gen implicado: HPRT (OMIM 308000) se localiza en Xq26.2-q26.3
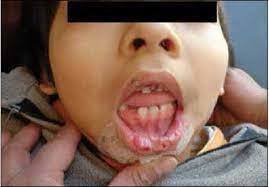
- Síndrome de Lesch-Nyhan (Deficiencia completa) Frecuencia de 1/235 000 a 1/380 000 RN vivos, cursa con hiperuricemia e hiperuricosuria, Ts. motores cognoscitivos y de comportamiento con automutilación y anemia megaloblástica
- El síndrome de Kelly-Seegmiller (deficiencia parcial) Presencia de gota y sus complicaciones pero sin involucramiento neurológico
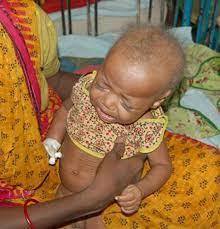
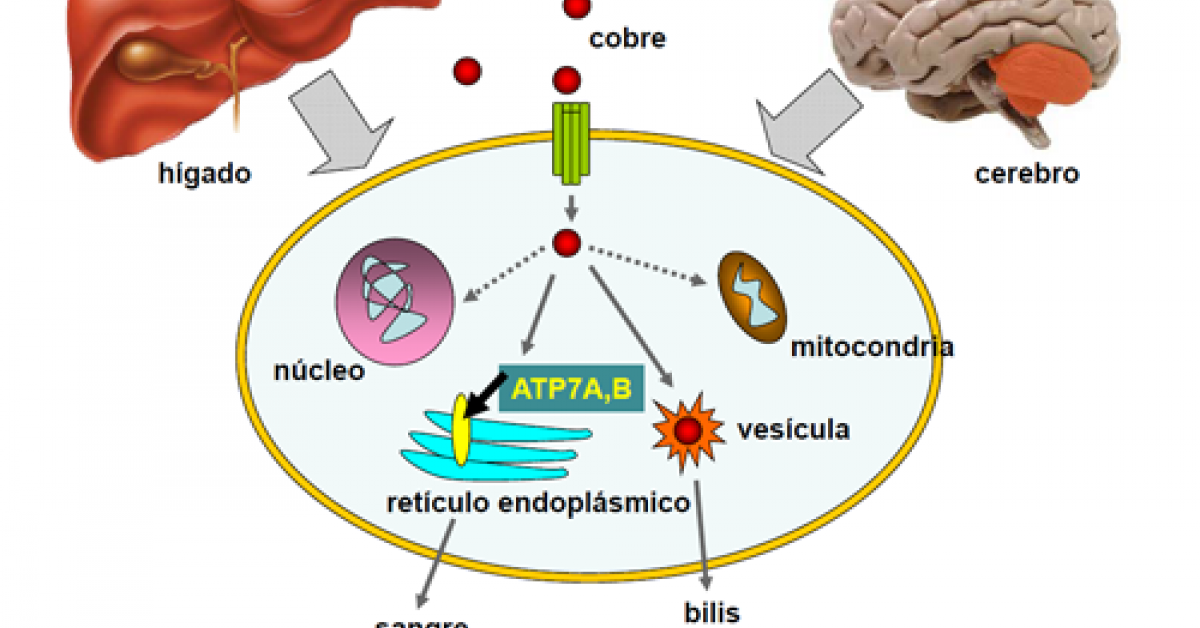
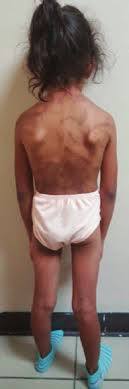
- SÍNDROME DE MENKES (SM)
- Trastorno neurodegenerativo por deficiencia en el metabolismo del cobre. Frecuencia de 1 en 50 000 a 1 en 250 000 nacimientos
- Gen de Menkes: (ATP7A) se localiza en Xq13.3
- Fenotipo clínico
- Forma clásica. Embarazo normal (17% son prematuros con peso <2 500 g) Nacimiento: hipotermia, hipoglucemia e ictericia, sin embargo, en la mayoría no se detectan alteraciones durante los dos primeros meses
- Craneofacial: pelo, corto en zona posterior y laterales, opaco, tieso, hipopigmentado, frágil y torcido. Dato patognomónico:pili torti
- Facies: cejas y pestañas escasas, puente nasal deprimido, mejillas caídas y papada marcada, el paladar es alto y retraso en erupción dentaria
- Síndrome del cuerno occipital.Variante alélica que tiene concentraciones séricas de cobre y ceruloplasmina bajas. Predomina afección en el tejido conectivo y en el óseo
- Heterocigocidad con mutaciones recesivas: 50% de sus células expresarán el alelo de la enfermedad , 50% el alelo normal
- PADECIMIENTOS
- El cobre es requerido en el metabolismo energético, biosíntesis de catecolaminas y formación del tejido conectivo.
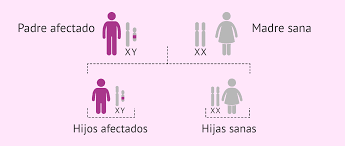
- HERENCIA DOMINANTE LIGADA AL CROMOSOMA X
- Menos frecuentes El individuo sólo necesita heredar una copia del gen mutado localizado en el cromosoma X para estar afectado.
- Mujer Portadora + varón sano= 50% de riesgo de descendencia afectada Genotípicamente las mujeres serán heterocigotas y los varones hemicigotos
- Varón afectado + mujer sana= Todas sus hijas heredarán el gen y serán heterocigotas afectadas Todos sus varones no heredarán el gen de la enfermedad (sanos)
- PADECIMIENTOS
- SÍNDROME DE ALPORT
- Alteración renal hereditaria de inicio posnatal con una prevalencia estimada de 1:50 000, debida a la alteración de la membrana basal glomerular por defecto en las cadenas de la colágena tipo IV
- Seis cadenas diferentes: COL4A1–COL4A2 en el cromosoma 13 COL4A3-COL4A4 en el cromosoma 2q35-37 COL4A5- COL4A6 en Xq22-23
- Forma de transmisión más frecuente: Ligada al X (80-85% de casos) resultado de mutaciones en la cadena α5
- Cuadro clínico de expresividad variable, manifestaciones Renales: Hematuria (signo inicial más frecuente), Proteinuria, Hipertensión
- Tratamiento eficaz: trasplante renal
- 3 a 4% de los casos desarrollan el Sx. de Goodpasture
- Padecimientos mortales en el varón
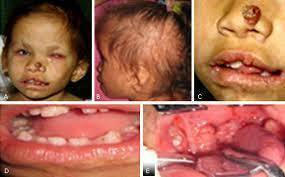
- INCONTINENTIA PIGMENTI
- Sx. neurocutáneo que presenta una constelación de manifestaciones en tejidos derivados del ectodermo y neuroectodermo que condicionan afección a nivel dermatológico, neurológico, oftalmológico y dental.
- Gen involucrado: NEMO/IKKgamma con locus en Xq28 el cual interviene en la activación del factor de transcripción NF-kB
- Cuadro clínico Piel y anexos:Etapa 1 o vesicular, Etapa 2 o verrucosa, Etapa 3 o hiperpigmentada, Etapa 4 o atrofica/hipopigmentada
- Criterios diagnósticos:
- Criterios mayores - Lesiones vesiculares neonatales con eosinofilia. - Hiperpigmentación en líneas de Blaschko que disminuyen en la adolescencia. - Lesiones atróficas lineales.
- Criterios menores - Alteraciones dentales. - Alopecia. - Pelo hirsuto y ensortijado. - Distrofia ungueal.
- HIPOPLASIA DÉRMICA FOCAL O SX. DE GOLTZ
- Caracterizado por anomalías cutáneas y una gran variedad de alteraciones en diversos órganos y sistemas
- Rasgos característicos: Femenino 90%, Osteopatía estriada, Coloboma, Ausencia de elementos derivados ectoneuromesodermo, Deformidad en pinzas de cangrejo
- Gen involucrado: PORCN localizado en Xp11.23, que interviene con las proteínas de señalización Wnt claves en el desarrollo embrionario
- MANIFESTACIONES CLÍNICAS Inspección general: talla baja, asimetría corporal y hombros caídos. Cuadro muy grave: mueren en los primeros años de vida En la historia familiar se registra alta incidencia de abortos o mortinatos
- HERENCIA LIGADA AL CROMOSOMA Y (HOLÁNDRICA)
- Herencia de los genes que se localizan en el cromosoma Y, estará presente sólo en varones (transmisión estricta padre-hijo) y nunca la transmitirá un varón a sus hijas
- Contiene genes implicados en la espermatogénesis, el gen que inicia la diferenciación sexual en embriones XY y un antígeno secundario de histocompatibilidad denominado HY.
- No hay transmisión varón a varón Todas sus hijas recibirán el cromosoma X mutado del padre y serán portadoras
- Formas leves
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
- ELABORADO POR: NALLELY GALLEGOS SÁNCHEZ 6°C BIBLIOGRAFÍA: Del Castillo Ruiz Victoria, Uranga Hernández Rafael Dulijh, Z. de la R. G. (2012). Génetica Clinica (Primera). Manual Moderno
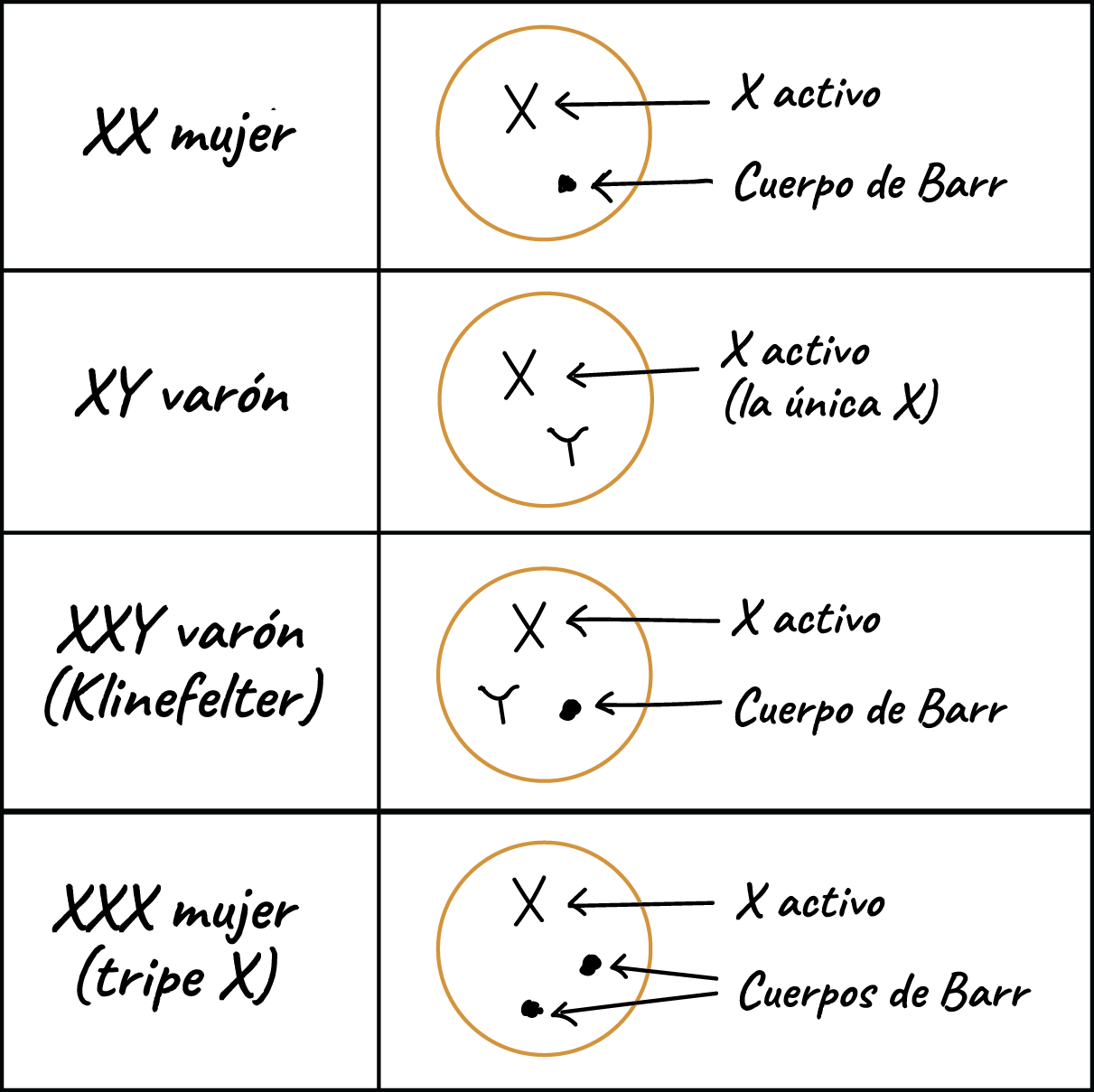{kind=link}
Want to create your own Flowcharts for free with GoConqr? Learn more.