6687527
Glomeruloesclerosis focal y
segmentaria
- Concepto/Definición
- Es una enfermedad
caracterizada por cicatrices
glomerulares segmentarias en
algunos glomérulos y se
manifiesta en forma de
proteinuria
- Esclerosis de algunos glomérulos
aunque no de todos (focal) y en los
glomérulos afectados solo se altera una
porción del ovillo capilar (segmentaria)
- Cuando se presenta por
primera vez es habitual
la hipertensión,
hematuria microscópica y
azoemia
- Esclerosis de algunos glomérulos
aunque no de todos (focal) y en los
glomérulos afectados solo se altera una
porción del ovillo capilar (segmentaria)
- Es una enfermedad
caracterizada por cicatrices
glomerulares segmentarias en
algunos glomérulos y se
manifiesta en forma de
proteinuria
- Demografía
- Principal causa de síndrome
nefrótico en adultos (33%)
- Población más afectada:
Hispanos y afroamericanos
- Principal causa de síndrome
nefrótico en adultos (33%)
- Impacto
- La prevalencia de GSFS primaria en
pacientes negros es 2-4 veces mayor
que en blancos y es más frecuente en
hombres (10-20%) y en niños (7-15%) (3)
- Representa la causa más frecuente
de glomerulonefritis primaria (3)
- Constituye la causa
más frecuente de
síndrome nefrótico (3)
- La prevalencia de GSFS primaria en
pacientes negros es 2-4 veces mayor
que en blancos y es más frecuente en
hombres (10-20%) y en niños (7-15%) (3)
- Etiología
- Enfermedad primaria (idiopática) 10-35%
- En asociación con enfermedades como: VIH,
adicción a heroína, obesidad masiva o
enfermedad de celulas falciformes
- Secundario a cicatrización por lesiones
previamente activas
- Nefropatía IgA
- Nefropatía IgA
- Componente de una respuesta adaptativa
- A pérdida de tejido renal: Ablación real
- Anomalías congénitas: Agenesia o displasia
- Adquirida: Nefropatía por reflujo
- A pérdida de tejido renal: Ablación real
- Formas hereditarias del síndrome nefrótico
por mutaciones en genes de proteínas en el
diafragma en hendidura
- Podocina
- α-actinina 4
- Podocina
- Enfermedad primaria (idiopática) 10-35%
- Fisiopatología
- Degeneración y alteración focal de
células epiteliales y endoteliales
con borramiento de podocitos
- Causado por aumento de la permeabilidad a proteínas, regulado por linfocitos T
- Defectos genéticos
- Anormalidades de
podocitos
- Anormalidades de
podocitos
- Proliferación celular (de células mesangiales, infiltrado de neutrófilos
- Esclerosis (regulada por TGF-B) inicialmente segmentaria y finalmente global
- Causado por aumento de la permeabilidad a proteínas, regulado por linfocitos T
- Hialinosis y esclerosis
- Por el atrapamiento de
proteínas plasmáticas en focos
hiperpermeables y aumento del
depósito en la MEC
- Por el atrapamiento de
proteínas plasmáticas en focos
hiperpermeables y aumento del
depósito en la MEC
- Degeneración y alteración focal de
células epiteliales y endoteliales
con borramiento de podocitos
- Clínica
- Hematuria
- Descenso del FG e hipertensión
- Proteinuria no selectiva
- Respuesta insuficiente a corticoesteroides
- Progreso a nefropatía crónica y en 50%
a nefropatía terminal
- Hematuria
- Auxiliares
diagnósticos
- Laboratorio: Proteinuria en rango nefrótico,
hematuria y en algunos casos hay
aumento de creatinina sérica y BUN (3)
- Laboratorio: Proteinuria en rango nefrótico,
hematuria y en algunos casos hay
aumento de creatinina sérica y BUN (3)
- Genética
- Gen NPHS1: Codifica nefrina, componente
del diafragma en hendidura
- Síndrome nefrótico congénito que produce
glomerulopatía de cambios mínimos
- Síndrome nefrótico congénito que produce
glomerulopatía de cambios mínimos
- GEFS autosómica recesiva: mutación de
NPHS2 que codifica podocina
- Síndrome nefrótico resistente a
corticoesteroides en la infancia
- Síndrome nefrótico resistente a
corticoesteroides en la infancia
- Proteína α actinina 4 de unión a la
actina de los podocitos
- GEFS autosómica dominante de
inicio insidioso, con alta
progresión a insuficiencia renal
- GEFS autosómica dominante de
inicio insidioso, con alta
progresión a insuficiencia renal
- Gen que codifica TRPC6 en los podocitos
- Gen NPHS1: Codifica nefrina, componente
del diafragma en hendidura
- Morfología
macro
- inicialmente las lesiones afectan a los glomérulos
yuxtamedulares, posteriormente se hacen más generalizadas
- inicialmente las lesiones afectan a los glomérulos
yuxtamedulares, posteriormente se hacen más generalizadas
- Morfología
micro
- Microscopio óptico
- En segmentos escleróticos: Colapso de asas capilares
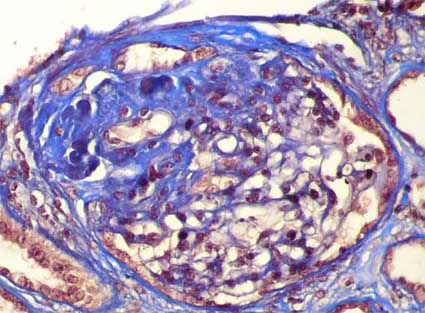
- Glomérulo con esclerosis en la
mitad superior teñida en azul,
la mitad inferior presenta una
arquitectura conservada
- Glomérulo con esclerosis en la
mitad superior teñida en azul,
la mitad inferior presenta una
arquitectura conservada
- Aumento de la matriz con depositos de proteínas
plasmáticas a lo largo de la pared capilar (hialinosis)
que pueden ocluir las luces capilares
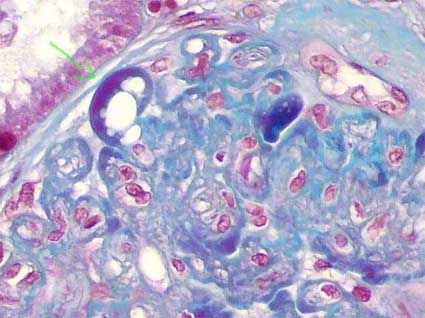
- Imagen con tricrómico de Masson donde se
observa hialinosis segmentaria con
vacuolas lipídicas, observaadas en color
rojizo. En las zonas adyacentes hay
disminución o pérdida de las luces capilares.
- Imagen con tricrómico de Masson donde se
observa hialinosis segmentaria con
vacuolas lipídicas, observaadas en color
rojizo. En las zonas adyacentes hay
disminución o pérdida de las luces capilares.
- Pueden existir gotas lipídicas y células espumosas
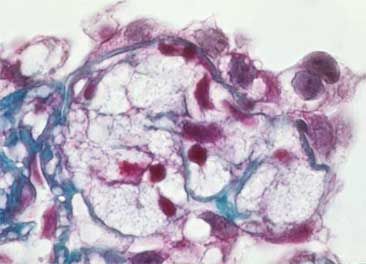
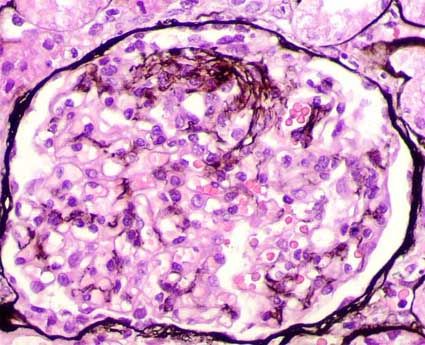
- Imagen de un glomérulo teñido
con tricromico de Masson donde se
observan células espumosas, son
células grandes con citoplasma
claro vacuolado y pequeños núcleos.
- Fernández Pacheco Miriam. Grupo: 3631
20/10/2016
- Fernández Pacheco Miriam. Grupo: 3631
20/10/2016
- Imagen de un glomérulo teñido
con tricromico de Masson donde se
observan células espumosas, son
células grandes con citoplasma
claro vacuolado y pequeños núcleos.
- En segmentos escleróticos: Colapso de asas capilares
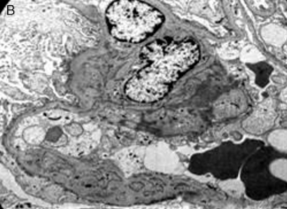
- Microscopio electrónico
- Borramiento difuso de los podocitos y puede
haber desprendimiento focal de células epiteliales
- Imagen de un área glomerular con fusión
pedicular, incremento de matriz
mesangial y depósitos electrodensos
- Imagen de un área glomerular con fusión
pedicular, incremento de matriz
mesangial y depósitos electrodensos
- Borramiento difuso de los podocitos y puede
haber desprendimiento focal de células epiteliales
- Microscopio óptico
- Métodos
especiales
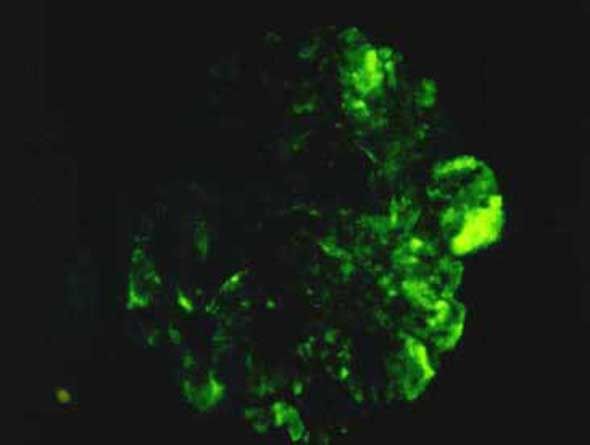
- Inmunofluorescencia
- Se puede observar IgM y
C3 en las áreas
escleróticas y/o mesangio
- Depósitos focales y segmentarios de IgM
y C3, que representan, en su mayoría
atrapamiento de proteínas plasmáticas
en las lesiones esclerosantes o hialinas
- Depósitos focales y segmentarios de IgM
y C3, que representan, en su mayoría
atrapamiento de proteínas plasmáticas
en las lesiones esclerosantes o hialinas
- Se puede observar IgM y
C3 en las áreas
escleróticas y/o mesangio
- Tinciones especiales
- Segmentos escleróticos (colágena IV)
positivos a PAS y plata metenamina
- Segmentos escleróticos (colágena IV)
positivos a PAS y plata metenamina
- Inmunofluorescencia
- Tratamiento
- FSGS primaria: Inhibidores del SRAA (2)
- Px con proteinuria en
límites nefróticos
pueden ser tratados
con corticoesteroides (2)
- FSGS secundaria: tratar la
causa básica y controlar la
proteinuria. No son útiles los
corticoesteroides ni otros
inmunosupresores (2)
- FSGS primaria: Inhibidores del SRAA (2)
- Pronóstico
- No hay remisión
espontánea
- 20% presentan evolución rápida (2 años) a
insuficiencia renal, con proteinuria masiva intratable
- Factores asociados a progresión rápida:
Gravedad de la proteinuria, de la insuficiencia
renal y subtipo histológico
- Bibliografía: 1. Kumar, V., Abbas, A., & Aster, J.. (2015). Robbins y Cotran. Patología Estructural y Funcional. 9ª edición,
Barcelona, España: Elsevier.// 2.. Dennis Kasper, & Anthony Fauci. Harrison. Principios de Medicina Interna, 19e. Mc Graw
Hill// 3. Avendaño H. Nefrología Clínica. 3e. Panamericana // 4. Vazquez H. Orientación diagnóstica de enfermedades renales
glomerulares. Revista española de patología. Vol. 46 N.1 2013. Elsevier
- Bibliografía: 1. Kumar, V., Abbas, A., & Aster, J.. (2015). Robbins y Cotran. Patología Estructural y Funcional. 9ª edición,
Barcelona, España: Elsevier.// 2.. Dennis Kasper, & Anthony Fauci. Harrison. Principios de Medicina Interna, 19e. Mc Graw
Hill// 3. Avendaño H. Nefrología Clínica. 3e. Panamericana // 4. Vazquez H. Orientación diagnóstica de enfermedades renales
glomerulares. Revista española de patología. Vol. 46 N.1 2013. Elsevier
- No hay remisión
espontánea
Media attachments
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Want to create your own Mind Maps for free with GoConqr? Learn more.