25372913
Descripción
Mapa Mental por Daniel Cacuango, actualizado hace más de 1 año
|
|
Creado por Daniel Cacuango
hace alrededor de 4 años
|
|
FARMACOLOGÍA
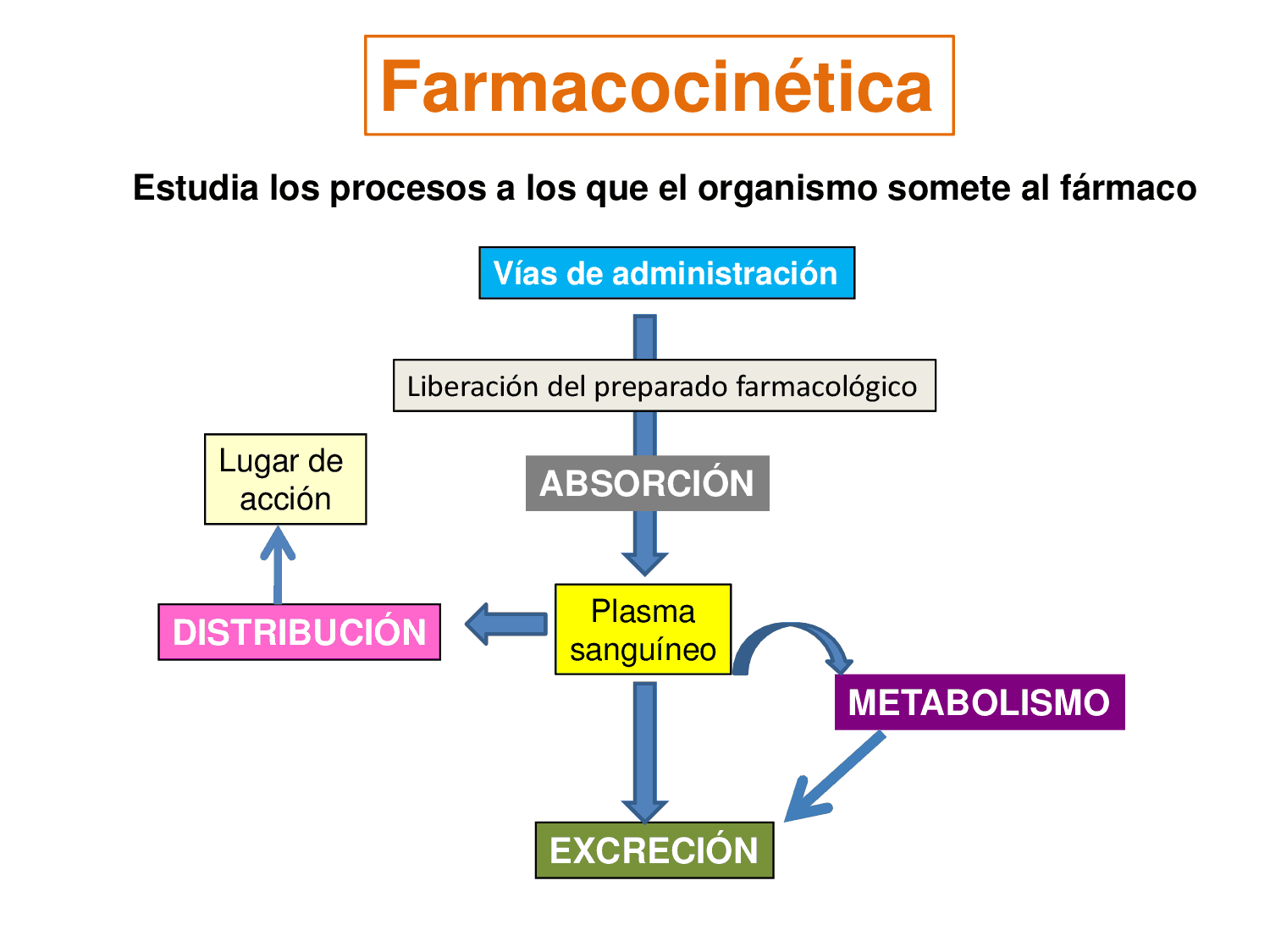
- Farmacocinética
- Generalidades
- Describe las acciones de una sustancia activa contenida en un
medicamento sobre el organismo una vez se ha ingerido o
administrado.
- Describe las acciones de una sustancia activa contenida en un
medicamento sobre el organismo una vez se ha ingerido o
administrado.
- Vía de administración
del fármaco
- Enteral
- Oral
- Los medicamentos orales son fácilmente administrados, las toxicidades, sobredosis o ambas
pueden solucionarse fácilmente con antídotos, como el carbón activado, además esta vía puede
ser más complicada, debido a que, el bajo pH gástrico inactiva algunos medicamentos.
- Preparaciones
orales
- Preparaciones con cubierta entérica: protege el medicamento del ácido gástrico,
llevándolo al intestino (menos ácido), donde el recubrimiento se disuelve y libera el
fármaco. Es útil para ácido-lábiles y fármacos irritantes para el estómago.
- Preparaciones de liberación extendida: tienen recubrimientos o ingredientes que controlan
la liberación del fármaco, lo que permite una absorción más lenta y una duración más
prolongada de la acción. Pueden administrarse a dosis menos frecuentes y mejorar así el
cumplimiento del paciente. Sirven para los fármacos con vida media breve.
- Preparaciones con cubierta entérica: protege el medicamento del ácido gástrico,
llevándolo al intestino (menos ácido), donde el recubrimiento se disuelve y libera el
fármaco. Es útil para ácido-lábiles y fármacos irritantes para el estómago.
- Preparaciones
orales
- Los medicamentos orales son fácilmente administrados, las toxicidades, sobredosis o ambas
pueden solucionarse fácilmente con antídotos, como el carbón activado, además esta vía puede
ser más complicada, debido a que, el bajo pH gástrico inactiva algunos medicamentos.
- Sublingual/bucal
- La vía sublingual se coloca debajo de la lengua y la bucal entre el carrillo y la encía. Ambas
vías incluyen su facilidad de administración, absorción rápida, derivación del ambiente
gastrointestinal agresivo y evitación del metabolismo de primer paso.
- La vía sublingual se coloca debajo de la lengua y la bucal entre el carrillo y la encía. Ambas
vías incluyen su facilidad de administración, absorción rápida, derivación del ambiente
gastrointestinal agresivo y evitación del metabolismo de primer paso.
- Oral
- Parenteral
- Se administra fármacos directamente en la circulación
sistémica. Se usa para fármacos que se absorben de
forma deficiente en las vías gastrointestinales o
inestables en las vías gastrointestinales, para
pacientes que no pueden tomar medicamentos por vía
oral (pacientes inconscientes) y en casos que requiere
inicio rápido de la acción. La administración puede
causar dolor, miedo, daño local a los tejidos e
infecciones.
- Intravenosa (IV): permite un efecto rápido y un grado máximo de control sobre la cantidad de
fármaco administrada. Cuando se inyecta en forma de bolo, la cantidad se administra a la
circulación casi de inmediato. En una infusión IV, el fármaco se infunde a lo largo de periodos
más prolongados para una mayor duración del fármaco circulante.
- Intramuscular (IM): los fármacos pueden estar en soluciones acuosas, que se absorben con
rapidez, o en preparaciones de depósito especializadas, que se absorben lentamente.
- Subcutánea (SC): la inyección SC proporciona absorción a través de difusión simple. La SC minimiza riesgos
de hemólisis o trombosis relacionados con la IV y puede proporcionar efectos constantes, lentos y
sostenidos. No se usa con fármacos que causan irritación tisular, ya que provocan dolor intenso y necrosis.
- Intradérmica (ID): consiste en la inyección a la dermis, se administra en agentes para determinación
diagnóstica y desensibilización.
- Intravenosa (IV): permite un efecto rápido y un grado máximo de control sobre la cantidad de
fármaco administrada. Cuando se inyecta en forma de bolo, la cantidad se administra a la
circulación casi de inmediato. En una infusión IV, el fármaco se infunde a lo largo de periodos
más prolongados para una mayor duración del fármaco circulante.
- Se administra fármacos directamente en la circulación
sistémica. Se usa para fármacos que se absorben de
forma deficiente en las vías gastrointestinales o
inestables en las vías gastrointestinales, para
pacientes que no pueden tomar medicamentos por vía
oral (pacientes inconscientes) y en casos que requiere
inicio rápido de la acción. La administración puede
causar dolor, miedo, daño local a los tejidos e
infecciones.
- Otros
- Inhalación oral y preparaciones nasales: tanto la inhalación oral como las vías nasales de
administración permiten suministrar con rapidez un fármaco a lo largo de una gran área de
superficie de membranas mucosas de las vías respiratorias y el epitelio pulmonar.
- Intratecal/intraventricular: Se administra directamente en el líquido cefalorraquídeo. cuando
se requiere de efectos locales y rápidos.
- Tópica: se usa cuando se busca un efecto local del fármaco.
- Transdérmica: se aplica medicamentos a la piel, mediante un parche transdérmico. La
velocidad de absorción varía dependiendo las características físicas de la piel, en el sitio de
aplicación y de la liposolubilidad del fármaco.
- Rectal: es útil para prevenir la destrucción del fármaco en el ambiente gastrointestinal o si
este induce el vómito al administrar por vía oral y si el paciente está inconsciente. La
absorción rectal suele ser errática e incompleta y muchos fármacos irritan la mucosa rectal.
- Inhalación oral y preparaciones nasales: tanto la inhalación oral como las vías nasales de
administración permiten suministrar con rapidez un fármaco a lo largo de una gran área de
superficie de membranas mucosas de las vías respiratorias y el epitelio pulmonar.
- Enteral
- Diseño y optimización del
esquema de dosificación
- Esquemas de infusión continua
- Concentración plasmática de un fármaco después de infusión IV continua: cuando se inicia
una infusión IV continua, la concentración plasmática del fármaco se eleva hasta alcanzar
un estado estable. La constante de velocidad para alcanzar un estado estable es la
constante de velocidad para la eliminación corporal total del fármaco
- Concentración plasmática de un fármaco después de infusión IV continua: cuando se inicia
una infusión IV continua, la concentración plasmática del fármaco se eleva hasta alcanzar
un estado estable. La constante de velocidad para alcanzar un estado estable es la
constante de velocidad para la eliminación corporal total del fármaco
- Esquemas de dosis fija/tiempo fijo
- Inyecciones IV múltiples: cuando un fármaco se administra de forma repetida a intervalos
regulares (menores de 5 vidas medias), la concentración plasmática aumenta hasta que se
alcanza un estado estable y se eliminan de forma exponencial con el tiempo.
- Administraciones orales múltiples: son la mayoría de los fármacos administrados de forma
ambulatoria, se toman a una dosis específica una, dos o más veces al día. Se absorben
lentamente y la concentración plasmática se ve influida por la velocidad de absorción y
eliminación.
- Inyecciones IV múltiples: cuando un fármaco se administra de forma repetida a intervalos
regulares (menores de 5 vidas medias), la concentración plasmática aumenta hasta que se
alcanza un estado estable y se eliminan de forma exponencial con el tiempo.
- Optimización de la
dosis
- Dosis de mantenimiento: los fármacos se administran para mantener una Css dentro de
la ventana terapéutica. Toma 4 a 5 vidas medias para que un fármaco alcance una Css .
- Dosis de carga: se administra para que el fármaco alcance la concentración plasmática deseada con rapidez,
seguida por una dosis de mantenimiento para mantener el estado estable. En general, la dosis de carga
puede calcularse como Dosis de carga = (Vd ) × (concentración plasmática deseada en estado estable)/F
- Ajuste de la dosis: se debe conocer los principios farmacocinéticos para ajustar dosis
que optimicen el tratamiento en un paciente determinado.
- Dosis de mantenimiento: los fármacos se administran para mantener una Css dentro de
la ventana terapéutica. Toma 4 a 5 vidas medias para que un fármaco alcance una Css .
- Esquemas de infusión continua
- Absorción de
fármacos
- Mecanismos de absorción de fármacos a
partir de la vía gastrointestinal
- Difusión pasiva: es cuando la fuerza que impulsa el fármaco se mueve de un área de alta concentración a una de menor concentración.
- Difusión facilitada: es cuando las proteínas transportadoras sufren cambios conformacionales, lo que permite el paso de fármacos o
moléculas endógenas en el interior de las células.
- Transporte activo: es capaz de mover los fármacos contra un gradiente de concentración de una región de baja concentración
farmacológica a una de mayor concentración. El proceso es saturable y dependiente de energía.
- Endocitosis y exocitosis: se usa para transportar fármacos de un tamaño excepcionalmente grande a través de la membrana celular. La
endocitosis es el transporte de partículas y sustancias al interior de la célula, mientras que la exocitosis es lo ontrario.
- Difusión pasiva: es cuando la fuerza que impulsa el fármaco se mueve de un área de alta concentración a una de menor concentración.
- . Factores que influyen sobre la
absorción
- Efecto del pH sobre la absorción del fármaco: la mayoría de los fármacos son ácidos débiles o bases débiles,
Sin embargo, un fármaco pasa a través de las membranas con mayor facilidad si no ha sufrido cambios.
- Flujo de sangre al sitio de absorción: los intestinos reciben mucho más flujo sanguíneo que el estómago.
- Área de superficie total disponible para absorción: depende de si la superficie es rica en borde en cepillo.
- Tiempo de contacto en la superficie de absorción: Depende de la rapidez o retraso que sufra un fármaco al transportarse.
- Expresión de glucoproteína P: en áreas de alta expresión, esta reduce la absorción de fármacos.
- Efecto del pH sobre la absorción del fármaco: la mayoría de los fármacos son ácidos débiles o bases débiles,
Sin embargo, un fármaco pasa a través de las membranas con mayor facilidad si no ha sufrido cambios.
- Bioequivalencia y otros tipos de equivalencia
- Dos formulaciones farmacológicas son bioequivalentes si muestran una biodisponibilidad comparable y
tiempos similares para alcanzar concentraciones sanguíneas máximas.
- La equivalencia terapéutica requiere que los principios activos sean bioequivalentes y
farmacéuticamente equivalentes.
- Dos formulaciones farmacológicas son bioequivalentes si muestran una biodisponibilidad comparable y
tiempos similares para alcanzar concentraciones sanguíneas máximas.
- Biodisponibilidad
- Determinación de biodisponibilidad: se determina al comparar las concentraciones plasmáticas del
fármaco después de una vía particular de administración
- Factores que influyen sobre la biodisponibilidad:
- Metabolismo hepático de primer paso: Si el fármaco se metaboliza con rapidez en el hígado o en el intestino
durante este paso inicial, la cantidad de fármaco sin cambio que entra a la circulación sistémica disminuye.
- Solubilidad del fármaco: Para que un fármaco se absorba con facilidad, debe ser altamente lipofílico,
pero con cierta solubilidad en soluciones acuosas.
- Inestabilidad química: algunos fármacos son inestables en el pH de los contenidos gástricos o se destruyen
en las vías gastrointestinales por enzimas degradativas.
- Naturaleza de la formulación farmacológica: la absorción del fármaco puede verse alterada por factores
que no están relacionados con la química del fármaco.
- Metabolismo hepático de primer paso: Si el fármaco se metaboliza con rapidez en el hígado o en el intestino
durante este paso inicial, la cantidad de fármaco sin cambio que entra a la circulación sistémica disminuye.
- Determinación de biodisponibilidad: se determina al comparar las concentraciones plasmáticas del
fármaco después de una vía particular de administración
- Mecanismos de absorción de fármacos a
partir de la vía gastrointestinal
- Depuración del fármaco por el
riñón
- Los pacientes con disfunción
renal pueden ser incapaces
de excretar fármacos y están
en riesgo de acumulación de
fármaco y efectos adversos.
- Eliminación renal de un
fármaco
- Filtración glomerular: los fármacos entran al riñón a través de las arterias renales, que se dividen
para formar un plexo capilar glomerular. El fármaco libre (no unido a albúmina) fluye a través de
las hendiduras capilares en el espacio de Bowman como parte del filtrado glomerular.
- Secreción tubular proximal: los fármacos que no se transfirieron en el filtrado glomerular dejan
los glomérulos a través de las arteriolas eferentes, que se dividen para formar un plexo capilar
que rodea la luz néfrica en el túbulo proximal.
- Reabsorción tubular distal pasiva: a medida que un fármaco se mueve hacia el túbulo
contorneado distal, su concentración aumenta y excede la del espacio perivascular.
- Filtración glomerular: los fármacos entran al riñón a través de las arterias renales, que se dividen
para formar un plexo capilar glomerular. El fármaco libre (no unido a albúmina) fluye a través de
las hendiduras capilares en el espacio de Bowman como parte del filtrado glomerular.
- Eliminación renal de un
fármaco
- Los pacientes con disfunción
renal pueden ser incapaces
de excretar fármacos y están
en riesgo de acumulación de
fármaco y efectos adversos.
- Depuración farmacológica del
metabolismo
- Las tres vías principales de eliminación son el metabolismo hepático, la
eliminación biliar y la excreción urinaria.
- Los procesos de eliminación disminuyen la concentración
plasmática de forma exponencial.
- La depuración (CL) se calcula: CL = 0.693 × Vd /t1/2
- Cinética del metabolismo: la transformación metabólica de los fármacos es
catalizada por enzimas, a esto se le llama cinética de primer paso. Por otra parte en
la cinética de orden cero, la enzima es saturada por una elevada concentración de
fármaco libre y la velocidad del metabolismo permanece constante con el tiempo.
- Reacciones del metabolismo del fármaco: El riñón no puede excretar de forma
eficiente los fármacos lipofílicos que cruzan con facilidad las membranas
celulares y se reabsorben en los túbulos contorneados distales.
- Estos agentes se metabolizan mediante dos fases: Fase 1
(convierten fármacos lipofílicos en moléculas más polares al
introducir o desenmascarar un grupo funcional polar) y la
Fase 2 (consiste de reacciones de conjugación).
- Estos agentes se metabolizan mediante dos fases: Fase 1
(convierten fármacos lipofílicos en moléculas más polares al
introducir o desenmascarar un grupo funcional polar) y la
Fase 2 (consiste de reacciones de conjugación).
- Reacciones del metabolismo del fármaco: El riñón no puede excretar de forma
eficiente los fármacos lipofílicos que cruzan con facilidad las membranas
celulares y se reabsorben en los túbulos contorneados distales.
- Cinética del metabolismo: la transformación metabólica de los fármacos es
catalizada por enzimas, a esto se le llama cinética de primer paso. Por otra parte en
la cinética de orden cero, la enzima es saturada por una elevada concentración de
fármaco libre y la velocidad del metabolismo permanece constante con el tiempo.
- La depuración (CL) se calcula: CL = 0.693 × Vd /t1/2
- Los procesos de eliminación disminuyen la concentración
plasmática de forma exponencial.
- Las tres vías principales de eliminación son el metabolismo hepático, la
eliminación biliar y la excreción urinaria.
- Distribución
farmacológica
- Flujo de sangre
- Proceso mediante el cual un fármaco deja de forma
reversible el torrente sanguíneo y entra al líquido
extracelular y a los tejidos.
- La velocidad del flujo de sangre a los capilares tisulares varía ampliamente.
- La velocidad del flujo de sangre a los capilares tisulares varía ampliamente.
- Proceso mediante el cual un fármaco deja de forma
reversible el torrente sanguíneo y entra al líquido
extracelular y a los tejidos.
- Permeabilidad capilar
- Se determina por la estructura capilar (varía en términos de la fracción de la membrana basal) y por la
naturaleza química del fármaco.
- Se determina por la estructura capilar (varía en términos de la fracción de la membrana basal) y por la
naturaleza química del fármaco.
- Unión de fármacos a las proteínas
plasmáticas y los tejidos
- Unión a proteínas plasmáticas: la unión reversible aísla los fármacos en una forma no difundible y hace más
lenta la transferencia fuera del compartimiento vascular. La albúmina es la principal proteína de unión.
- Unión a las proteínas tisulares: Los fármacos se acumulan por la unión a lípidos, proteínas o ácidos
nucleóticos. Los reservorios tisulares sirven como fuente mayor del fármaco y prolongan sus acciones o
causan toxicidad farmacológica local.
- Unión a proteínas plasmáticas: la unión reversible aísla los fármacos en una forma no difundible y hace más
lenta la transferencia fuera del compartimiento vascular. La albúmina es la principal proteína de unión.
- Lipofilicidad
- Los fármacos lipofílicos se mueven con facilidad a través de la mayoría de las membranas biológicas.
Estos fármacos se disuelven en las membranas lípidas y penetran la totalidad de la superficie celular.
- Los fármacos lipofílicos se mueven con facilidad a través de la mayoría de las membranas biológicas.
Estos fármacos se disuelven en las membranas lípidas y penetran la totalidad de la superficie celular.
- Volumen de
distribución
- Se calcula al dividir la dosis que eventualmente llega a la
circulación sistémica entre la concentración plasmática al tiempo
cero (C0 ).
- Distribución en los compartimientos de agua en el
cuerpo
- Una vez que el fármaco entra al cuerpo, tiene
el potencial de distribuirse en cualquiera de
los tres compartimientos funcionales.
- Compartimiento plasmático
- Líquido extracelular
- Agua corporal total
- Compartimiento plasmático
- Una vez que el fármaco entra al cuerpo, tiene
el potencial de distribuirse en cualquiera de
los tres compartimientos funcionales.
- Determinación del Vd: Se calcula dividiendo la dosis
sobre la concentración plasmática al tiempo cero.
- Efecto del Vd en la vida media del fármaco: cualquier factor que aumenta el
Vd puede aumentar la vida media y extender la duración de la acción del
fármaco.
- Distribución en los compartimientos de agua en el
cuerpo
- Se calcula al dividir la dosis que eventualmente llega a la
circulación sistémica entre la concentración plasmática al tiempo
cero (C0 ).
- Flujo de sangre
- Excreción por otras
vías
- Los fármacos que no se absorben después de la administración oral de fármacos
que se secretan directamente en el intestino o en la bilis se excretan en las
heces.
- La eliminación de fármacos en la leche materna puede exponer al lactante que
está alimentándose.
- La depuración corporal total y la vida media farmacológica son
medidas importantes de la depuración farmacológica que se usan
para optimizar el tratamiento farmacológico y minimizar la toxicidad.
- La depuración corporal total y la vida media farmacológica son
medidas importantes de la depuración farmacológica que se usan
para optimizar el tratamiento farmacológico y minimizar la toxicidad.
- La excreción de la mayoría de los fármacos en el sudor, la saliva, las lágrimas,
el pelo y la piel solo ocurre en un grado reducido.
- Los fármacos que no se absorben después de la administración oral de fármacos
que se secretan directamente en el intestino o en la bilis se excretan en las
heces.
- Generalidades
- Interacciones
farmaco-receptor y
farmacodinamia
- Generalidades
- Describe las acciones y los efectos, tanto dañinos como benéficos, de un
fármaco en el organismo. El complejo fármaco-receptor es una interacción
fisicoquímica que se produce entre diferentes moléculas al admnistrar un
fármaco.
- Describe las acciones y los efectos, tanto dañinos como benéficos, de un
fármaco en el organismo. El complejo fármaco-receptor es una interacción
fisicoquímica que se produce entre diferentes moléculas al admnistrar un
fármaco.
- Actividad
intrínseca
- Determina la
capacidad para
activar de forma
parcial o total los
receptores.
- Agonistas totales
- Es cuando el fármaco se une al receptor y produce una respuesta biológica máxima que simula la
respuesta al ligando endógeno, estos a la vez se unen a un receptor, estabilizan el receptor en su estado
activo y se dice que tienen una actividad intrínseca de uno. En los agonistas totales las curvas de
dosis-respuesta para la unión al receptor y cada una de las respuestas biológicas deben ser comparables.
- Es cuando el fármaco se une al receptor y produce una respuesta biológica máxima que simula la
respuesta al ligando endógeno, estos a la vez se unen a un receptor, estabilizan el receptor en su estado
activo y se dice que tienen una actividad intrínseca de uno. En los agonistas totales las curvas de
dosis-respuesta para la unión al receptor y cada una de las respuestas biológicas deben ser comparables.
- Agonistas parciales
- Tienen actividades intrínsecas mayores de cero pero menores de uno, al no producir el
mismo Emáx suele actuar como un agonista parcial de un agonista total, por lo que suele
ser considerada como utilidad terapéutica.
- Tienen actividades intrínsecas mayores de cero pero menores de uno, al no producir el
mismo Emáx suele actuar como un agonista parcial de un agonista total, por lo que suele
ser considerada como utilidad terapéutica.
- Agonistas inversos
- Tienen una actividad intrínseca menor de cero, revierten el estado de activación de los
receptores y ejercen el efecto farmacológico opuesto de los agonistas. Además estabilizan la
forma R inactiva y hacen que R* se convierta en R.
- Tienen una actividad intrínseca menor de cero, revierten el estado de activación de los
receptores y ejercen el efecto farmacológico opuesto de los agonistas. Además estabilizan la
forma R inactiva y hacen que R* se convierta en R.
- Antagonista
- No tiene efecto sobre la
función biológica en ausencia
de un agonista, pero puede
disminuir su efecto.
- Antagonistas competitivos: si el antagonista se une al mismo sitio en el
receptor que el agonista en una forma reversible, es “competitivo”.
- Antagonistas irreversibles: causa una desviación hacia debajo de Emáx , sin cambio de los valores EC50 y se
unen de forma covalente al sitio activo del receptor, reduciendo los receptores disponibles del agonista.
- Antagonistas alostéricos: se une a un sitio distinto al de la unión agonista y previene la activación del
receptor. Además cambia una desviación debajo de la Emáx de un agonista, sin cambiar el valor EC50 .
- Antagonismo funciona o fisiológico: actúa en un receptor completamente separado, iniciando efectos
funcionalmente opuestos a los del agonista.
- Antagonistas competitivos: si el antagonista se une al mismo sitio en el
receptor que el agonista en una forma reversible, es “competitivo”.
- No tiene efecto sobre la
función biológica en ausencia
de un agonista, pero puede
disminuir su efecto.
- Agonistas totales
- Determina la
capacidad para
activar de forma
parcial o total los
receptores.
- Transducción de
señal
- Características de la transducción de
señal
- Tiene dos
características
importantes
- Amplificación de señal: los receptores ligados a proteína G y enzimas tienen la capacidad
de amplificar la intensidad de señal y la duración mediante el efecto de cascada de señal.
- Desensibilización y regulación negativa de los receptores: la administración continua de un agonista o
antagonista produce cambios en la capacidad de respuesta del receptor. El receptor puede
desensibilizarse debido a demasiada estimulación agonista, resultando en una respuesta disminuida.
- Amplificación de señal: los receptores ligados a proteína G y enzimas tienen la capacidad
de amplificar la intensidad de señal y la duración mediante el efecto de cascada de señal.
- Tiene dos
características
importantes
- El complejo
fármaco-receptor
- Las células poseen diferentes de receptores, cada uno es específico para un agonista particular y
produce una respuesta única. La magnitud de la respuesta celular es proporcional al número de
complejos fármaco-receptor.
- Las células poseen diferentes de receptores, cada uno es específico para un agonista particular y
produce una respuesta única. La magnitud de la respuesta celular es proporcional al número de
complejos fármaco-receptor.
- Estados de
receptores
- Existen receptores en dos estados, inactivo (R) y activo (R*), están en equilibrio reversible entre sí,
por lo general favoreciendo el estado inactivo. , los agonistas, antagonistas y agonistas parciales son
moléculas que se unen al sitio de activación en el receptor y pueden afectar la fracción de R*.
- Existen receptores en dos estados, inactivo (R) y activo (R*), están en equilibrio reversible entre sí,
por lo general favoreciendo el estado inactivo. , los agonistas, antagonistas y agonistas parciales son
moléculas que se unen al sitio de activación en el receptor y pueden afectar la fracción de R*.
- Principales familias de
receptores
- Se dividen en cuatro familias: 1) canales iónicos con compuerta de ligandos, 2) receptores acoplados
a proteína G, 3) receptores ligados a enzimas y 4) receptores intracelulares.
- Se dividen en cuatro familias: 1) canales iónicos con compuerta de ligandos, 2) receptores acoplados
a proteína G, 3) receptores ligados a enzimas y 4) receptores intracelulares.
- Características de la transducción de
señal
- Relación dosis-respuesta
cuantal
- Es aquella entre la dosis del fármaco y la
proporción de una población de pacientes
que responden a él.
- Las curvas dosis-respuesta cuantales son útiles para
determinar las dosis a las que responde la mayoría de la
población.
- Índice terapéutico: es una medida de la seguridad del fármaco, el valor más grande indica
un amplio margen entre las dosis que son efectivas y aquellas que son tóxicas. Se calcula:
TI = TD50 /ED50 El IT
- Utilidad clínica del índice terapéutico
- Warfarina (fármaco con índice terapéutico pequeño): a medida que se aumenta la
dosis de warfarina, una mayor fracción de los pacientes responde. A mayores dosis ocurre
anticoagulación que resulta en hemorragia en un pequeño porcentaje de pacientes.
- Penicilina (fármaco con gran índice terapéutico): es seguro y frecuente administrar 86 dosis
en exceso para lograr una respuesta deseada sin el riesgo de efectos adversos. En este caso,
la biodisponibilidad no altera de forma crítica los efectos terapéuticos o clínicos.
- Warfarina (fármaco con índice terapéutico pequeño): a medida que se aumenta la
dosis de warfarina, una mayor fracción de los pacientes responde. A mayores dosis ocurre
anticoagulación que resulta en hemorragia en un pequeño porcentaje de pacientes.
- Utilidad clínica del índice terapéutico
- Índice terapéutico: es una medida de la seguridad del fármaco, el valor más grande indica
un amplio margen entre las dosis que son efectivas y aquellas que son tóxicas. Se calcula:
TI = TD50 /ED50 El IT
- Las curvas dosis-respuesta cuantales son útiles para
determinar las dosis a las que responde la mayoría de la
población.
- Es aquella entre la dosis del fármaco y la
proporción de una población de pacientes
que responden a él.
- Relaciones de dosis
respuesta
- Relación dosis
graduada-respuesta
- A medida que aumenta la concentración
de un fármaco, su efecto farmacológico
también aumenta de forma gradual
hasta que todos los receptores están
ocupados
- Dos importantes
características del
fármaco
- Potencia: es una medida de la cantidad del fármaco necesaria para producir un efecto.
- Eficacia: es la magnitud de respuesta que causa un fármaco cuando interactúa con un receptor.
- Potencia: es una medida de la cantidad del fármaco necesaria para producir un efecto.
- Dos importantes
características del
fármaco
- A medida que aumenta la concentración
de un fármaco, su efecto farmacológico
también aumenta de forma gradual
hasta que todos los receptores están
ocupados
- Efecto de la concentración del fármaco
sobre la unión a receptores
- Se aplica la ley de la Masa de acción, ya que, al asumir que la unión de una molécula del
fármaco no altera la unión de moléculas subsecuentes puede expresarse matemáticamente
la relación entre el porcentaje de receptores unidos y la concentración del fármaco.
- Se aplica la ley de la Masa de acción, ya que, al asumir que la unión de una molécula del
fármaco no altera la unión de moléculas subsecuentes puede expresarse matemáticamente
la relación entre el porcentaje de receptores unidos y la concentración del fármaco.
- Relación de la unión del fármaco con el
efecto farmacológico
- Se debe aplicar la ley de Acción de masa tomando en cuenta 3 suposiciones: 1) La
magnitud de la respuesta es proporcional a la cantidad de receptores ocupados por
el fármaco, 2) el Emáx ocurre cuando se unen todos los receptores y 3) una
molécula del fármaco se une a solo una molécula del recepto
- Se debe aplicar la ley de Acción de masa tomando en cuenta 3 suposiciones: 1) La
magnitud de la respuesta es proporcional a la cantidad de receptores ocupados por
el fármaco, 2) el Emáx ocurre cuando se unen todos los receptores y 3) una
molécula del fármaco se une a solo una molécula del recepto
- Relación dosis
graduada-respuesta
- Generalidades
- 1. Whalen, K.. Farmacología [Internet]. Barcelona (España): Wolters Kluwer; 2019 . Disponible en:
file:///C:/Users/59396/Downloads/TB1-2019%20LIR%20Farmacologia%207a%20Edicion%202019%20Whalen.pdf
Recursos multimedia adjuntos
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
¿Quieres crear tus propios Mapas Mentales gratis con GoConqr? Más información.