32017930
Descripción
Mapa Mental por meiby vega, actualizado hace más de 1 año
|
|
Creado por meiby vega
hace más de 3 años
|
|
Enfermedades genéticas que afectan
a la cavidad bucal
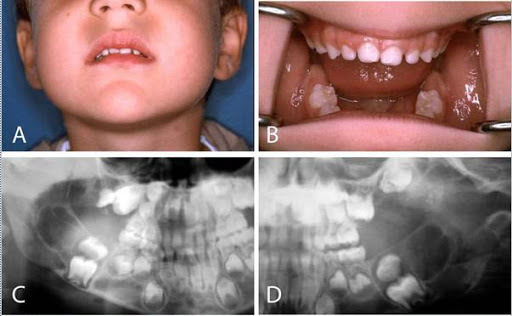
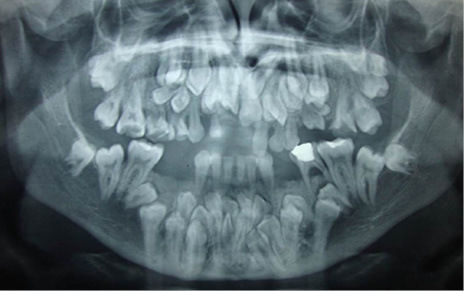
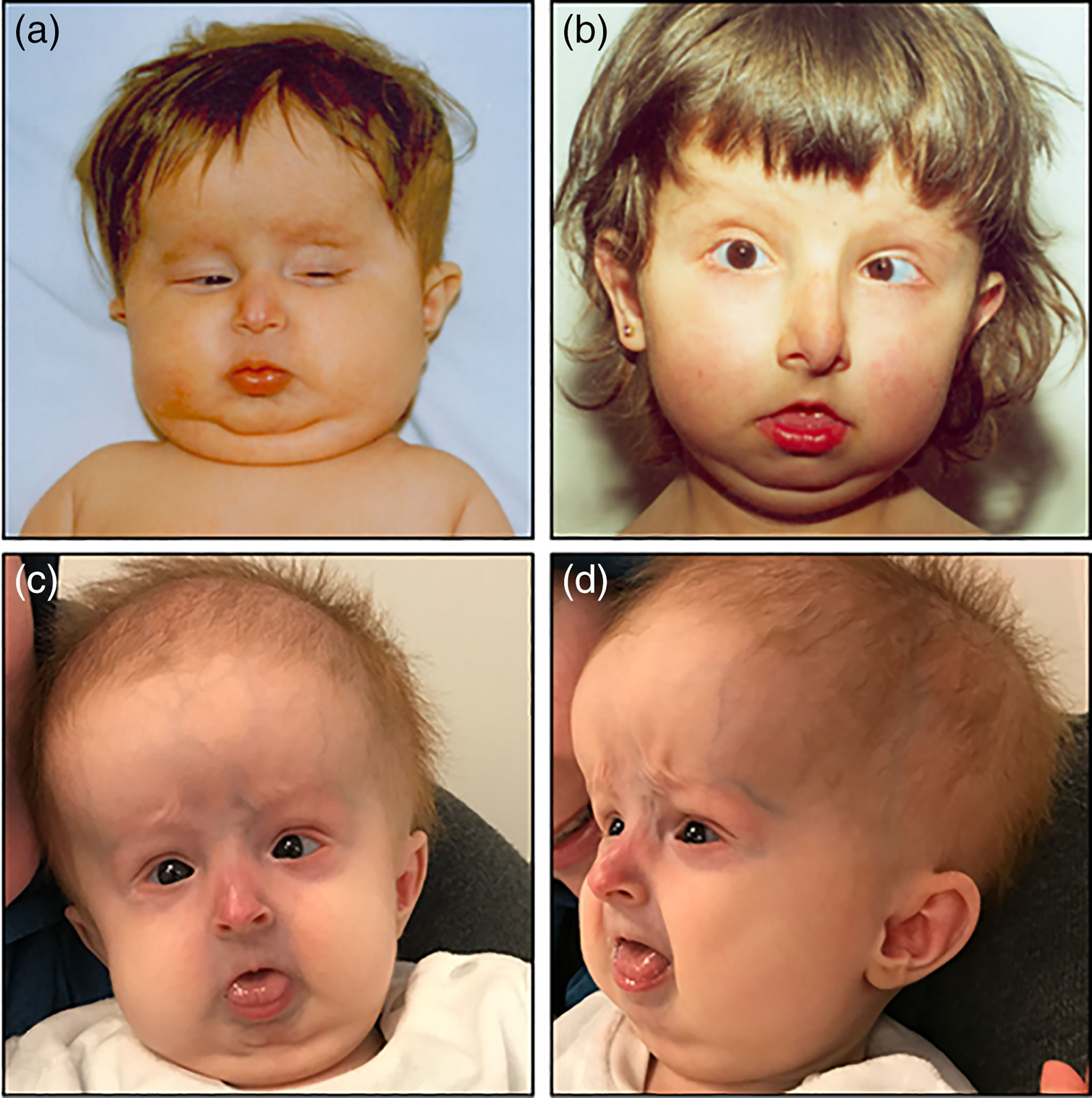
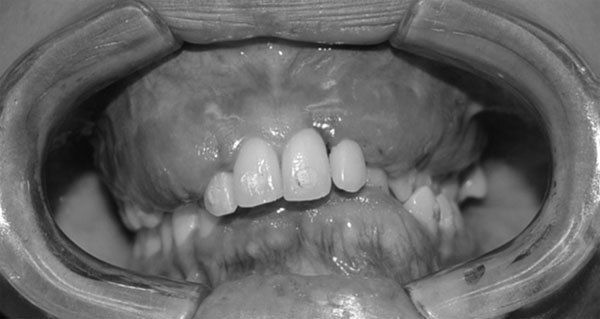
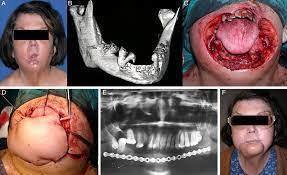
- Querubinismo
- OTROS NORMBRES: Displasia fibrosa familiar
de los maxilares, displasia fibrosa juvenil
entre otras
- Manifestaciones
clínicas
- se manifiesta en la infancia,
con frecuencia en la edad de los
3 a 4 años
- Los pacientes muestran una expansión
progresiva, no dolorosa, simétrica de los
maxilares da lugar a una cara sugestiva del
querubin
- Maxilares son duros a pala
palpación y puede haber
linfoadenopatías regionales
- La dentición primara se
puede exfoliar de manera
espontánea y prematura
- La mucosa bucal por lo
general esta intacta y de
color normal
- La mucosa bucal por lo
general esta intacta y de
color normal
- La dentición primara se
puede exfoliar de manera
espontánea y prematura
- Maxilares son duros a pala
palpación y puede haber
linfoadenopatías regionales
- Los pacientes muestran una expansión
progresiva, no dolorosa, simétrica de los
maxilares da lugar a una cara sugestiva del
querubin
- Etiología
- Se hereda como una enfermedad genpetica
autosómica dominante - Mutacioón del gen
SH3BP2 CROMOSOMA 4p16.3 AFECTA A LOS
MAXILARES CON EXPRESIVIDAD VARIABLE
- la penetración del gen
dominante es del 100% para el
sexo masculino y 70& para las
mujeres
- la penetración del gen
dominante es del 100% para el
sexo masculino y 70& para las
mujeres
- Manifestaciones
radriográficas
- Presentan radiograficamente una imagen radiolúcida
multilocular que produce destrucción bilateral del
huseo de uno o ambos maxilares con expansión y
adelgazamiento de las placas corticales
- En el maxilar inferior puede
afectar el cuerpo y la rama
produciendo perforación de la
corteza
- Los dientes
involucrados que no
han hecho erupción se
observan desplazados
- Los dientes
involucrados que no
han hecho erupción se
observan desplazados
- En el maxilar inferior puede
afectar el cuerpo y la rama
produciendo perforación de la
corteza
- Manifestaciones
histopatológicas
- El aspecto microscópico se caracteriza por la
presencia de células gigantes multinucleadas,
tejido conjuntivo fibrilar, fibroblastos, vasos
sanguíneos, células inflamatorias.
- En lesiones maduras el tejido
afectado se torna más fibroso y hay
una disminución en el número de
células gigantes
- En lesiones maduras el tejido
afectado se torna más fibroso y hay
una disminución en el número de
células gigantes
- El aspecto microscópico se caracteriza por la
presencia de células gigantes multinucleadas,
tejido conjuntivo fibrilar, fibroblastos, vasos
sanguíneos, células inflamatorias.
- Presentan radiograficamente una imagen radiolúcida
multilocular que produce destrucción bilateral del
huseo de uno o ambos maxilares con expansión y
adelgazamiento de las placas corticales
- Se hereda como una enfermedad genpetica
autosómica dominante - Mutacioón del gen
SH3BP2 CROMOSOMA 4p16.3 AFECTA A LOS
MAXILARES CON EXPRESIVIDAD VARIABLE
- se manifiesta en la infancia,
con frecuencia en la edad de los
3 a 4 años
- OTROS NORMBRES: Displasia fibrosa familiar
de los maxilares, displasia fibrosa juvenil
entre otras
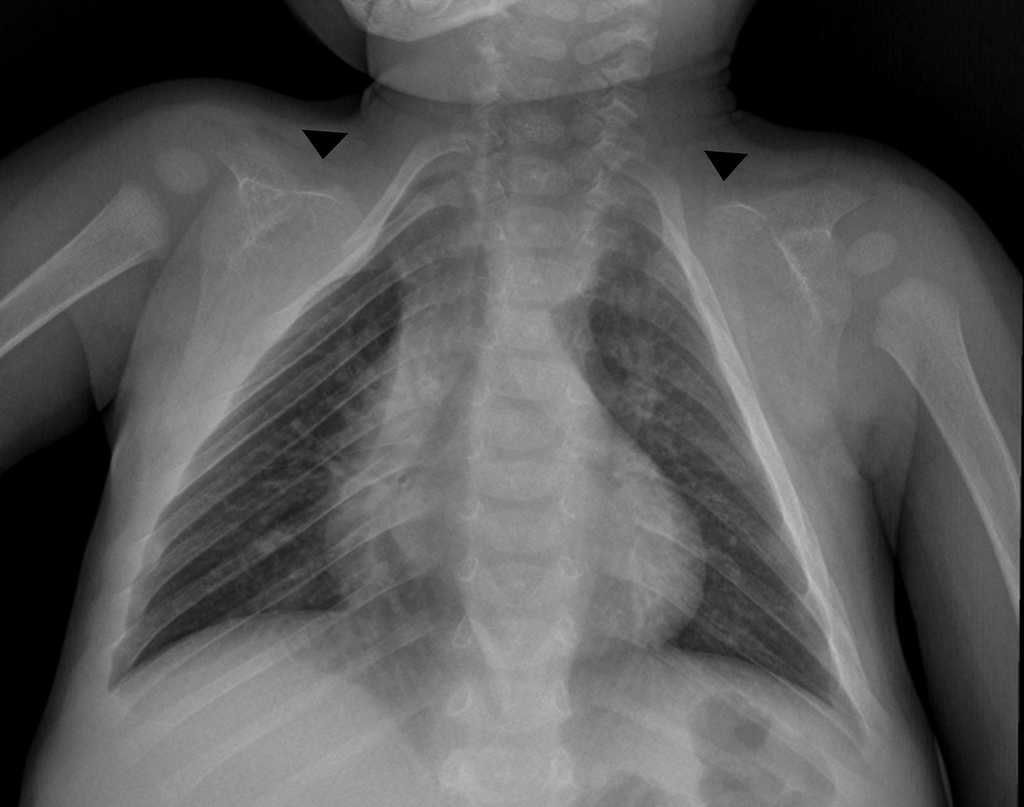
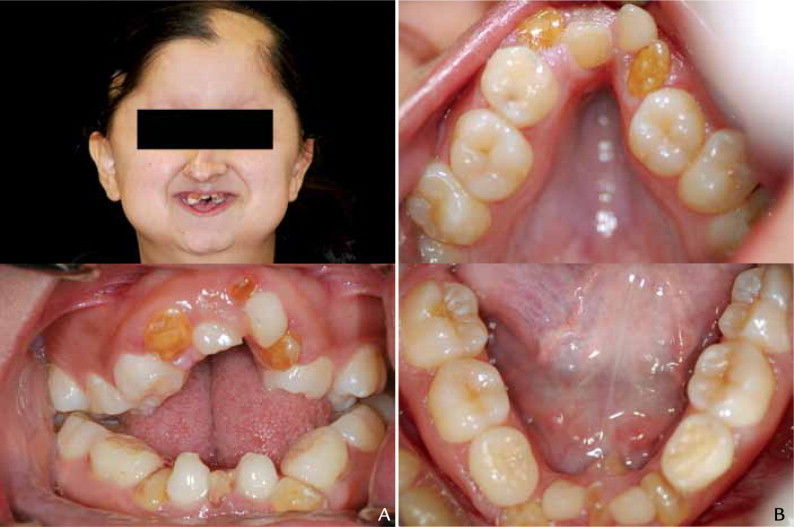
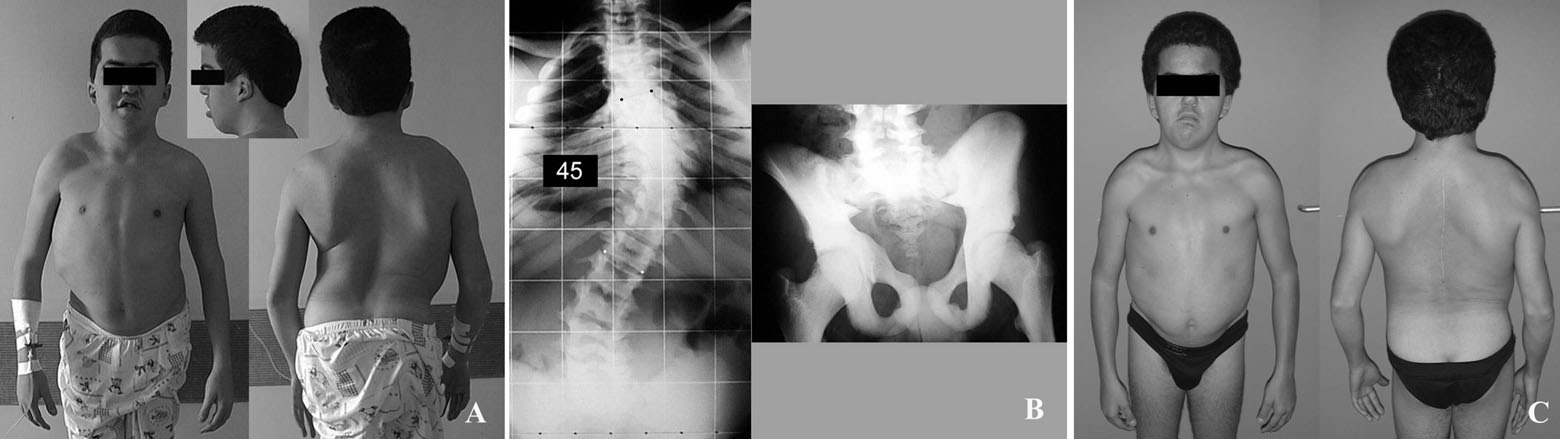
- Displasia
cleidocraneal
- Tambien llamada como:
Displasia cleidocraneal,
displasia osteodental,
enfermedad de Marie -
Sainton.
- se caracteriza por aplasia o
Hipoplasia de las clavículas,
malformaciones craneofaciales
características y la presencia de
gran número de dientes
supernumerarios no
erupcionados.
- se caracteriza por aplasia o
Hipoplasia de las clavículas,
malformaciones craneofaciales
características y la presencia de
gran número de dientes
supernumerarios no
erupcionados.
- Etiologia
- Se transmite por una
modalidad autosómica
dominante cpn alta
penetranicia y
expresividad variable
- Se debe por un brazo
corto de cromosoma
- Se debe por un brazo
corto de cromosoma
- Manifestaciones clínicas
- Craneofaciales: El cráneo es
braquicefálico, con un notable
abombamiento frontal y
parietal apariencia de cara
pequeña
- La nariz tiene la base ancha
y el puente nasal hundido.
Clavículas: A la palpación
puede observarse una
ausencia unilateral o bilateral
- Bucales: Las lesiones bucales
consisten en paladar ojival, Hipoplasia
maxilar que origina prognatismo
mandibular relativo, falta de unión de
la sínfisis mentoniana.
- Retraso de la resorción
fisiológica de la raíz de los
dientes primarios con
prolongada exfoliación de
los mismos.
- Bucales: Las lesiones bucales
consisten en paladar ojival, Hipoplasia
maxilar que origina prognatismo
mandibular relativo, falta de unión de
la sínfisis mentoniana.
- La nariz tiene la base ancha
y el puente nasal hundido.
Clavículas: A la palpación
puede observarse una
ausencia unilateral o bilateral
- Craneofaciales: El cráneo es
braquicefálico, con un notable
abombamiento frontal y
parietal apariencia de cara
pequeña
- Se transmite por una
modalidad autosómica
dominante cpn alta
penetranicia y
expresividad variable
- Tambien llamada como:
Displasia cleidocraneal,
displasia osteodental,
enfermedad de Marie -
Sainton.
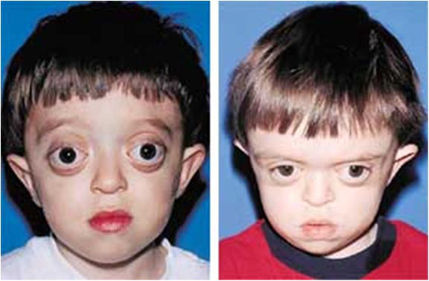
- Disostosis craneofacial
- Sinonimia:
Enfermedad de
Crouzon.
- alteraciones en la formación y
desarrollo del cráneo, Hipoplasia
maxilar, órbitas superficiales con
exoftalmos, estrabismo divergente.
- alteraciones en la formación y
desarrollo del cráneo, Hipoplasia
maxilar, órbitas superficiales con
exoftalmos, estrabismo divergente.
- Etiología y
patogenia
- se hereda con una
modalidad autosómica
dominante con
penetrancia completa y
expresividad variable
- se hereda con una
modalidad autosómica
dominante con
penetrancia completa y
expresividad variable
- Manifestaciones clínicas
- El labio
superior suele
ser corto y el
labio inferior
caído,
- Retardo en la
erupción dentaria
- Paladar
profundo y
ojival
- Erupción ectópica
de los primeros
molares
superiores
- Apiñamiento
maxilar
anterior
- Macroglosia relativa por
estrechez palatina
- Maloclusión
dentaria clase
III
- El labio
superior suele
ser corto y el
labio inferior
caído,
- Sinonimia:
Enfermedad de
Crouzon.
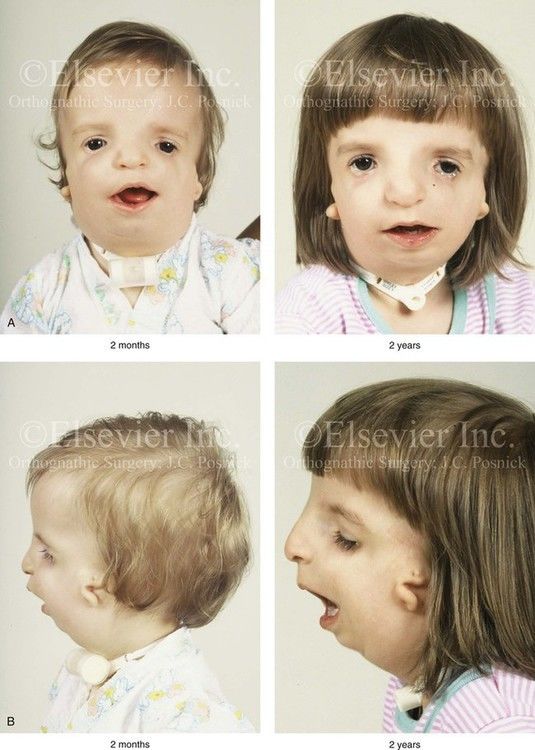
- Síndrome de Teacher Collins
- : Disostosis
mandibulo facial.
- Afecta principalmente las
estructuras en desarrollo del
primer arco branquial y en
menor grado el segundo arco
branquial.
- Afecta principalmente las
estructuras en desarrollo del
primer arco branquial y en
menor grado el segundo arco
branquial.
- Etiología y
patogenia
- Este sindrome se
transmite a partir de una
modalidad autosómica
dominate, algunos casos
se debe a mutación
espontánea
- Este sindrome se
transmite a partir de una
modalidad autosómica
dominate, algunos casos
se debe a mutación
espontánea
- Manifestaciones
clínicas
- Se observa Hipoplasia del
maxilar inferior, apófisis
cigomática del hueso temporal
y oídos externo y medio.
- • El 30% de los pacientes
muestran paladar
hendido
- Maloclusión dentaria,
dientes separados.
- Se observa Hipoplasia del
maxilar inferior, apófisis
cigomática del hueso temporal
y oídos externo y medio.
- : Disostosis
mandibulo facial.
- Síndrome de
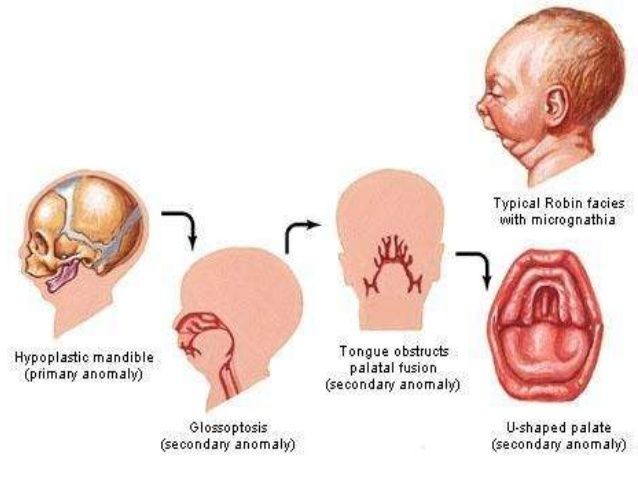
Pierre Robin
- Etiología y patogenia
- Se considera mala posición e interposición de la lengua entre las
placas del paladar durante el desarrollo fetal
- Puede ser causado por causas metabólicas
- Puede ser causado por causas metabólicas
- Se considera mala posición e interposición de la lengua entre las
placas del paladar durante el desarrollo fetal
- Manifestaciones clínicas
- Hipoplasia del maxilar inferior
- • Glosoptosis
- Dientes natales
- Dientes natales
- Lengua de apariencia
grande en relación
con la mandíbula
- • Glosoptosis
- Hipoplasia del maxilar inferior
- Etiología y patogenia
- Síndrome de
Hollermann -
Streiff
- Tambien llamado
Síndrome oculo -
mandíbulo - facial,
Discefalia oculo mandibulo
- facial, Discefalia con
catarata congénita e
hipotricosis.
- Etiología y patogenia
- Se transmite tanto en forma autosómica
recesiva como autosómica dominante, sin
embargo tambien puede ser una mutación de
NOVO
- Causa de la mutación es el gen GJA1
- Causa de la mutación es el gen GJA1
- Se transmite tanto en forma autosómica
recesiva como autosómica dominante, sin
embargo tambien puede ser una mutación de
NOVO
- Manifestaciones clínicas
- Micrognasia
- Microstomía
- Paladar ojival
- Ausencia dentaria
- Maloclusiones
- Malformaciones
dentarias
- Persistencia de
dientes
temporales
- Retraso en la
erupción de los
permanentes
- Dientes
supernumerarios
- Micrognasia
- Tambien llamado
Síndrome oculo -
mandíbulo - facial,
Discefalia oculo mandibulo
- facial, Discefalia con
catarata congénita e
hipotricosis.
- Osteopetrosis
- Sinonimia : Enfermedad de hueso
de mármol, enfermedad de Albers
- Schönberg, osteosclerosis fragilis
generalista.
- La osteopetrosis puede dividirse en tres grupos clínicos:, Osteopetrosis Maligna Infantil:, • Osteopetrosis
Autosómica Recesiva:, • Osteopetrosis Autosómica Dominante:
- Etiopatogenia
- la falta de resorción fisiológica
del hueso debido a la reducción de
la actividad osteoclástica
- los osteoclastos no responden de
manera apropiada a la presencia de
hormona paratiroidea o a estímulos
fisiológicos
- Los osteoclastos no sufren el
proceso llamado borde ondulado
y así permitir la liberación de
enzimas lisosomales en la
interfase célula - hueso.
- Los osteoclastos no sufren el
proceso llamado borde ondulado
y así permitir la liberación de
enzimas lisosomales en la
interfase célula - hueso.
- los osteoclastos no responden de
manera apropiada a la presencia de
hormona paratiroidea o a estímulos
fisiológicos
- la falta de resorción fisiológica
del hueso debido a la reducción de
la actividad osteoclástica
- Manifestaciones clínicas
- Retardo en el crecimiento
- Aumento de la
densidad ósea
junto con un
estado de
fragilidad
- Retraso en la
erupción dentaria
- Aumento de la
densidad ósea
junto con un
estado de
fragilidad
- Oclusión de la
cavidad
medular
- Falta de
resorción del
hueso alveolar
- Hipoplasia
del esmalte
- Prognatismo
mandibular
- Elevado índice de caries
secundarias a la
Hipoplasia del esmalte
- Tendencia a
desarrollar
osteomielitis
- Retardo en el crecimiento
- Sinonimia : Enfermedad de hueso
de mármol, enfermedad de Albers
- Schönberg, osteosclerosis fragilis
generalista.
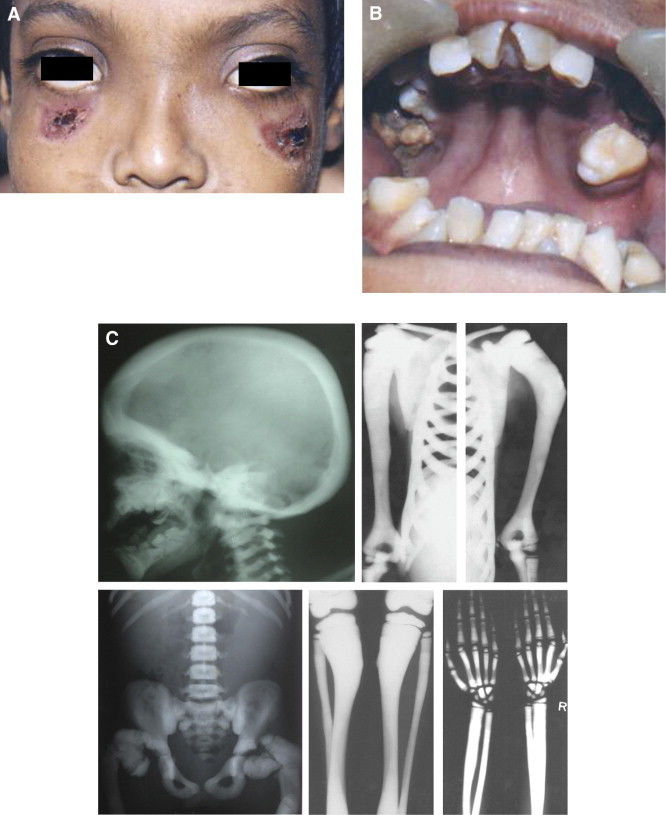
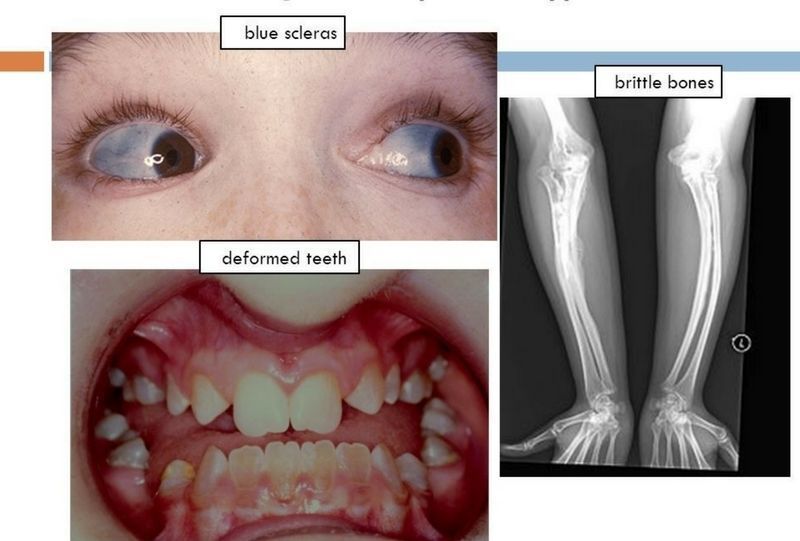
- Osteogénesis imperfecta
- Sinonimia: "Huesos frágiles,
Fragilitas ossium,
osteopsatirosis, Enfermedad de
Lobstein.
- comprende un grupo de trastornos
hereditarios generalizados del tejido
conjuntivo, con manifestaciones clínicas
en el esqueleto, oído, articulaciones,
ligamentos, dientes, esclerótica y piel
- existen 3 tipos
- Osteogénesis imperfecta tipo
I,Osteogénesis imperfecta tipo II,
Osteogénesis imperfecta tipo
III,Osteogénesis imperfecta tipo
IV
- Osteogénesis imperfecta tipo
I,Osteogénesis imperfecta tipo II,
Osteogénesis imperfecta tipo
III,Osteogénesis imperfecta tipo
IV
- existen 3 tipos
- comprende un grupo de trastornos
hereditarios generalizados del tejido
conjuntivo, con manifestaciones clínicas
en el esqueleto, oído, articulaciones,
ligamentos, dientes, esclerótica y piel
- Manifestaiones
- Osteogénesis imperfecta tipo I
- • La manifestación bucal
más común es la
Dentinogénesis
imperfecta produciendo
dientes mal formados y
de color azul amarillento
- Existe retardo en la erupción.
- Alta incidencia de
Maloclusión tipo II y
retención de molares.
- Alta incidencia de
Maloclusión tipo II y
retención de molares.
- Existe retardo en la erupción.
- • La manifestación bucal
más común es la
Dentinogénesis
imperfecta produciendo
dientes mal formados y
de color azul amarillento
- Osteogénesis
imperfecta tipo II
- • Es un síndrome letal y
la mitad de los fetos
afectados nacen
muertos.
- ausencia casi total
de osificación del
cráneo.
- s dentina atubular,
ausencia de predentina y
abundancia de fibras
argirófilas.
- s dentina atubular,
ausencia de predentina y
abundancia de fibras
argirófilas.
- ausencia casi total
de osificación del
cráneo.
- Osteogénesis imperfecta
tipo III
- Osteogénesis
imperfecta tipo III
- Es una afección
rara caracterizada
por fragilidad ósea
grave, fracturas
múltiples y
deformidad del
esqueleto.
- La Dentinogénesis
imperfecta y el retardo
en la erupción
- La Dentinogénesis
imperfecta y el retardo
en la erupción
- Es una afección
rara caracterizada
por fragilidad ósea
grave, fracturas
múltiples y
deformidad del
esqueleto.
- Osteogénesis
imperfecta tipo IV
- Es una osteopenia
hereditaria que causa
fragilidad de los
huesos
- A nivel bucal se
describen coronas
dentales cortas,
acampanadas y con
cuello estrecho
- Obliteración parcial o
total de la pulpa.
- Dentinogénesis
imperfecta relacionada
con Osteogénesis
imperfecta
- Maloclusión tipo II y
retención de molares.
- Maloclusión tipo II y
retención de molares.
- Dentinogénesis
imperfecta relacionada
con Osteogénesis
imperfecta
- Obliteración parcial o
total de la pulpa.
- A nivel bucal se
describen coronas
dentales cortas,
acampanadas y con
cuello estrecho
- Es una osteopenia
hereditaria que causa
fragilidad de los
huesos
- Osteogénesis
imperfecta tipo III
- • Es un síndrome letal y
la mitad de los fetos
afectados nacen
muertos.
- Osteogénesis imperfecta tipo I
- Sinonimia: "Huesos frágiles,
Fragilitas ossium,
osteopsatirosis, Enfermedad de
Lobstein.
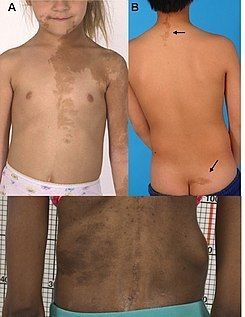
- Síndrome de Albright
- Sinonimia: Osteodistrofia
de Albright,
seudohipoparatiroidismo.
- es una enfermedad de
carácter dominante ligada al
cromosoma X.
- no hay respuesta en
el hueso y riñón a la
hormona
paratiroidea.
- no hay respuesta en
el hueso y riñón a la
hormona
paratiroidea.
- es una enfermedad de
carácter dominante ligada al
cromosoma X.
- Etiología
- Las
concentraciones
de
paratohormona
están
elevedas,
incluso
estando
el
paciente
hipocalcémico
- se debe a un transtorno
genético de los tejidos
especialmente riñon y
esqueleto, se transmiten
con un caracter
dominante ligado al
cromosomax
- se debe a un transtorno
genético de los tejidos
especialmente riñon y
esqueleto, se transmiten
con un caracter
dominante ligado al
cromosomax
- Las
concentraciones
de
paratohormona
están
elevedas,
incluso
estando
el
paciente
hipocalcémico
- Manifestaciones clpinicas
- El retardo en la erupción y la
Hipoplasia del esmalte
- : ápices abiertos,
Hipodoncia,
calcificaciones pulpares,
aplasia dental, paladar
ojival, cámaras
pulpares amplias.
- La hipoplasia merece
restauraciones dentales,
para las maloclusiones
se utiliza aparatos fijos
removibles
- La hipoplasia merece
restauraciones dentales,
para las maloclusiones
se utiliza aparatos fijos
removibles
- : ápices abiertos,
Hipodoncia,
calcificaciones pulpares,
aplasia dental, paladar
ojival, cámaras
pulpares amplias.
- El retardo en la erupción y la
Hipoplasia del esmalte
- Sinonimia: Osteodistrofia
de Albright,
seudohipoparatiroidismo.
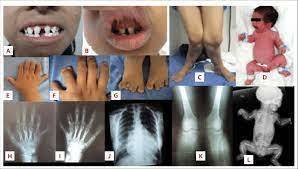
- Síndrome de Ellis - Van Creveld
- Sinonimia: Displasia
condroectodermica, displasia
mesoectodermal, enanismo
con seis dedos.
- genéticamente transmitida
con un patrón autosómico
recesivo, el cual involucra el
esqueleto, uñas, dientes.
- genéticamente transmitida
con un patrón autosómico
recesivo, el cual involucra el
esqueleto, uñas, dientes.
- Etiologia
- Existe
consanguinidad
de los padres
en una tercera
parte de los
casos.
- Existe
consanguinidad
de los padres
en una tercera
parte de los
casos.
- Manifestaciones
- Dientes neonatales
en el 25 % de los
casos
- Los dientes suelen
ser pequeños y
espaciados
- Hay retardo en la
erupción dentaria
permanente
- Las coronas dentarias
poseen formas
anómalas.
- • El esmalte es hipoplásico en
el 50% de los casos.
- • El esmalte es hipoplásico en
el 50% de los casos.
- Las coronas dentarias
poseen formas
anómalas.
- Hay retardo en la
erupción dentaria
permanente
- Los dientes suelen
ser pequeños y
espaciados
- Dientes neonatales
en el 25 % de los
casos
- Sinonimia: Displasia
condroectodermica, displasia
mesoectodermal, enanismo
con seis dedos.
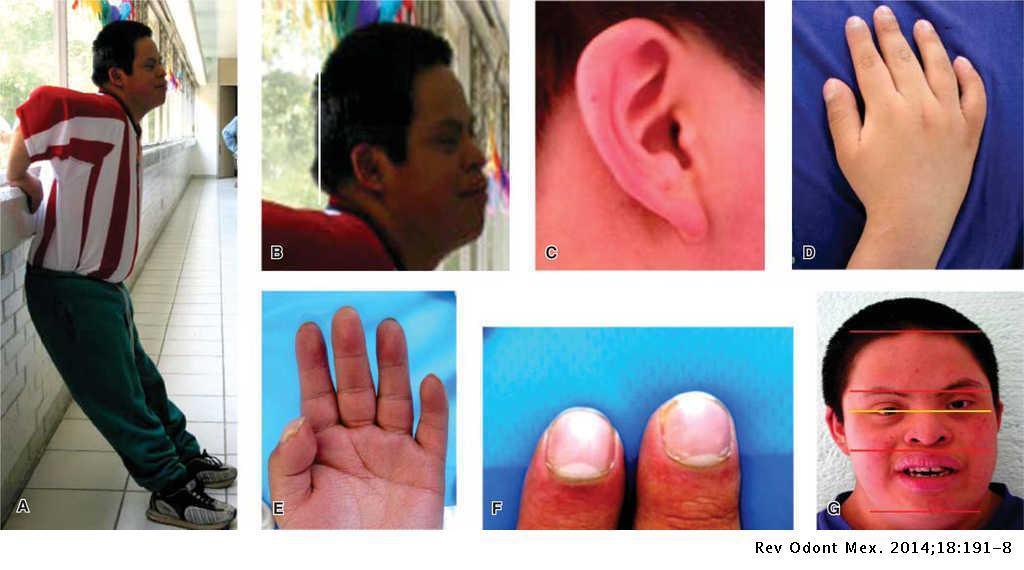
- Sindrome de down
- Sinonimia: Trisomía21.
- El síndrome de Down es una
aberración cromosómica
común y fácil de identificar
- El síndrome de Down es una
aberración cromosómica
común y fácil de identificar
- Etiología
- posibles causas
incluyen masaiquismo
no detectado en un
padre, exposición
repetida a un mismo
agresor ambiental,
progenitores de
cualquier edad ue
hayan tenido un hijo
con trisompia, poseen
un riesgo significativo
de tener otro hijo
afectado
- posibles causas
incluyen masaiquismo
no detectado en un
padre, exposición
repetida a un mismo
agresor ambiental,
progenitores de
cualquier edad ue
hayan tenido un hijo
con trisompia, poseen
un riesgo significativo
de tener otro hijo
afectado
- Manifestaciones clínicas
- Las manifestaciones bucales de
este síndrome incluyen: lengua
fisurada, macroglosia, protusión de
la lengua, úvula bífida, la erupción
de los dientes primarios y
permanentes se retrasa
- anormalidades de los dientes,
incluyendo corona y raíz,
hipocalcificación del esmalte,
prognatismo relativo y respiración
bucal.
- anormalidades de los dientes,
incluyendo corona y raíz,
hipocalcificación del esmalte,
prognatismo relativo y respiración
bucal.
- Las manifestaciones bucales de
este síndrome incluyen: lengua
fisurada, macroglosia, protusión de
la lengua, úvula bífida, la erupción
de los dientes primarios y
permanentes se retrasa
- Sinonimia: Trisomía21.
- Síndrome de múltiples
carcinomas basocelulares
nevoides y quistes de los
maxilares
- Sinonimia: Síndrome de
carcinoma nevoide de
células basales, síndrome
de Gorlin-Goltz.
- • Conjunto hereditario de
defectos múltiples en la piel,
sistema nervioso, ojos,
glándulas endocrinas y huesos
maxilares
- • Conjunto hereditario de
defectos múltiples en la piel,
sistema nervioso, ojos,
glándulas endocrinas y huesos
maxilares
- Etiología
- El sindrome de células
basales es heredado como
una característica
autosómica dominante
- como principal transtorno
es la aparicion de cáncer
de piel
- como principal transtorno
es la aparicion de cáncer
de piel
- El sindrome de células
basales es heredado como
una característica
autosómica dominante
- Manifestaciones
clpinicas
- Alteraciones dermatológica:
- • Alteraciones faciales:
Prognatismo mandibular,
queratoquistes odontogénicos
de los maxilares.
- Alteraciones oftalmológicas
- Alteraciones bucales:
Quistes mandibulares,
dentición anormal,
dientes retenidos
- Alteraciones esqueléticas:
- Alteraciones neurológicas
- Alteraciones neurológicas
- Alteraciones esqueléticas:
- Alteraciones bucales:
Quistes mandibulares,
dentición anormal,
dientes retenidos
- Alteraciones oftalmológicas
- • Alteraciones faciales:
Prognatismo mandibular,
queratoquistes odontogénicos
de los maxilares.
- Alteraciones dermatológica:
- Sinonimia: Síndrome de
carcinoma nevoide de
células basales, síndrome
de Gorlin-Goltz.
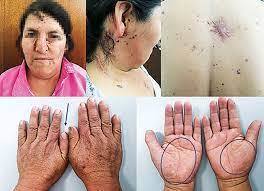
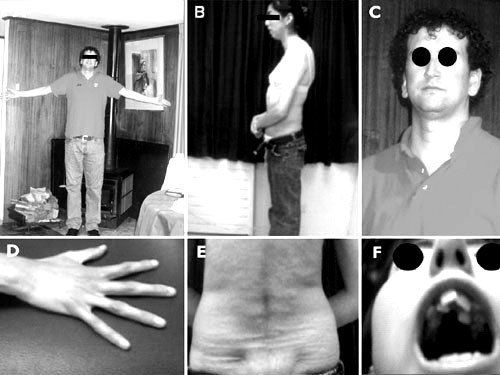
- Síndrome de marfan
- El síndrome de Marfan es una enfermedad
hereditaria del tejido conjuntivo
caracterizada por anomalías esqueléticas,
cardiovasculares y oculares.
- Etiología
- Deriva de una deficiencia en los enlances cruzados estables del coléno
- Se hereda de forma autosómica
dominante con un alto grado de
penetrancia y una expresividad
variable
- Deriva de una deficiencia en los enlances cruzados estables del coléno
- Manifestaciones clínicas
- Los pacienctes son delgados y de
estatura elevada, con brazos y piernas
relativamente largos, manos grandes
con dedos largos y articulaciones laxas.
- • El sistema cardiovascular suele
afectarse por dilatación de la
aorta (aneurismas).
- A nivel de la cavidad bucal
incluyen paladar ojival y
estrecho, dientes apiñados.
- • El sistema cardiovascular suele
afectarse por dilatación de la
aorta (aneurismas).
- Los pacienctes son delgados y de
estatura elevada, con brazos y piernas
relativamente largos, manos grandes
con dedos largos y articulaciones laxas.
- El síndrome de Marfan es una enfermedad
hereditaria del tejido conjuntivo
caracterizada por anomalías esqueléticas,
cardiovasculares y oculares.
Recursos multimedia adjuntos
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
¿Quieres crear tus propios Mapas Mentales gratis con GoConqr? Más información.