6685999
Descripción
Mapa Mental por Sofia Berdugo, actualizado hace 8 meses
|
|
Creado por Sofia Berdugo
hace alrededor de 8 años
|
|
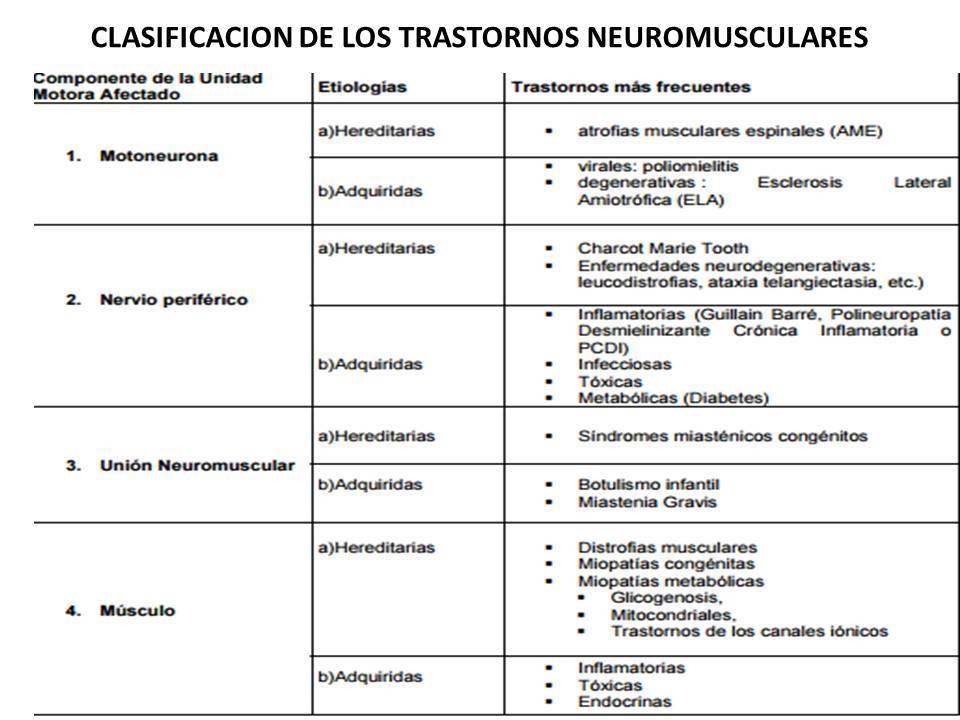
ENFERMEDADES NEUROMUSCULARES DE NERVIO PERIFÉRICO
- HEREDITARIAS
- Charcot-Marie-Tooth
atrofia muscular peroneal
- polineuropatía sensitivo-motora
- tipo
- desmielinizante o axonal
- desmielinizante o axonal
- tipo
- CAUSADA
- Mutación genetica
- Mutación genetica
- pérdida de visión por una
neuropatía óptica, parálisis de
cuerdas vocales o hipoacusia
neurosensorial
- TTO FT
- MOVILIZACIONES
- ALINEACIÓN
POSTURAL
- ejercicios
respiratorios
- Reeducación de la marcha
- ortesis
- ejercicios aeróbico y submaximos especialmente
en el curso temprano de la enfermad,
- evitar el sobreesfuerzo y trabajo lo cual
produciría más debilidad.
- Si hay dolor,
calambres
- modificar la
actividad
- modificar la
actividad
- Si hay dolor,
calambres
- estiramientos
- MOVILIZACIONES
- diagnostica
- Estudios de conducción nerviosa.
- Electromiografía.
- Biopsia del nervio.
- Análisis genéticos
- Estudios de conducción nerviosa.
- polineuropatía sensitivo-motora
- síntomas
- dificultad para correr,
pararse en los talones
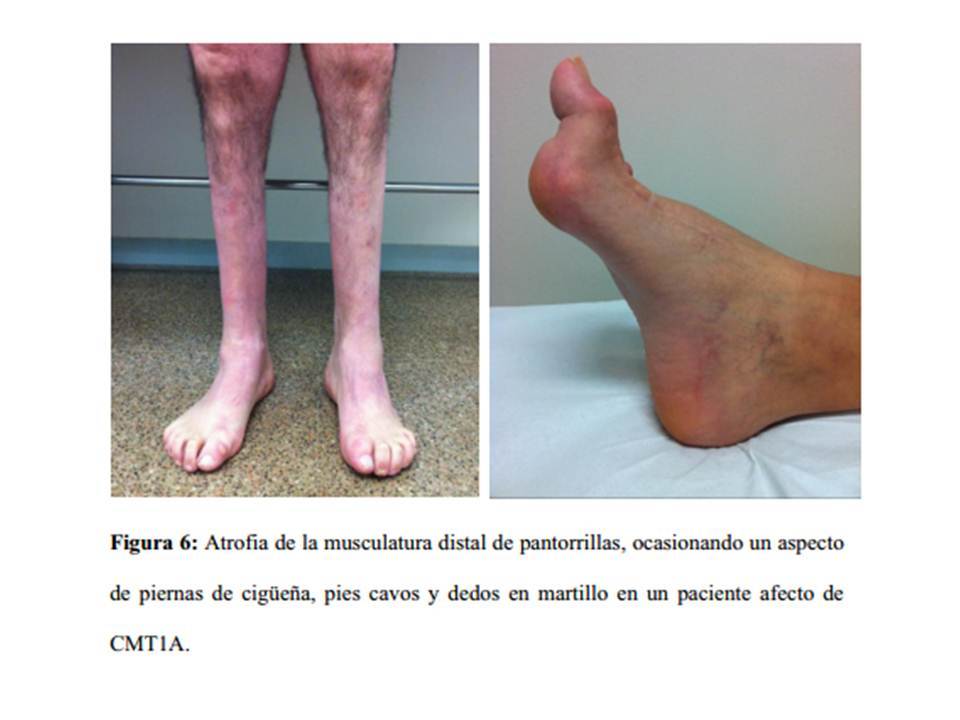
- atrofia
- inicialmente musc pie
- origina
- pie cavo y
dedos en
martillo
- luego
- los músculos de la pierna y el
tercio inferior del muslo
- marcha steppage
- marcha steppage
- los músculos de la pierna y el
tercio inferior del muslo
- luego
- pie cavo y
dedos en
martillo
- origina
- inicialmente musc pie
- disminución de la percepción
del dolor, temperatura o
propiocepción en miembros
inferiores
- mano en garra, temblor en manos,
calambres, pies fríos,
hiperqueratosis plantar y
acrocianosis
- dificultad para correr,
pararse en los talones
- tipos
- Tipo 1 (CMT1) o desmielinizante
- autosómica dominante
- gen en el cromosoma 17
- proteína 22 encargada de la mielina
- más adelante padecen de
debilidades en las manos y
la pérdida de sensación
- pacientes padecen de debilidad
- atrofia de los músculos inferiores de las
piernas desde la adolescencia
- más adelante padecen de
debilidades en las manos y
la pérdida de sensación
- proteína 22 encargada de la mielina
- gen en el cromosoma 17
- CMT1A neuropatía
hereditaria con predisposición a
parálisis compresiva
- disminución importante
- gen PMP-22
- neuropatías desmielinantes episódicas y recurrentes
- neuropatías desmielinantes episódicas y recurrentes
- gen PMP-22
- disminución importante
- CMT1B
- autosómica dominante
- mutación gen
- proteína cero (P0) de mielina
- clínica
- igual CMT!
- igual CMT!
- proteína cero (P0) de mielina
- mutación gen
- autosómica dominante
- autosómica dominante
- CMT2
- anormalidades
en el axón
- mutación en el gen1B-beta
- menor afectación
sensorial CMT1 mas
benigna
- menor afectación
sensorial CMT1 mas
benigna
- mutación en el gen1B-beta
- anormalidades
en el axón
- CMT3
- neuropatía DejerineSottas
(DJS)
- comienza en la infancia.
- atrofias y debilidades musculares severas
problemas sensoriales
- atrofias y debilidades musculares severas
problemas sensoriales
- comienza en la infancia.
- neuropatía DejerineSottas
(DJS)
- CMT4
- síntomas de debilidad en las
piernas durante la niñez y
pueden perder la capacidad
de caminar en la
adolescencia
- Escoliosis
- Escoliosis
- síntomas de debilidad en las
piernas durante la niñez y
pueden perder la capacidad
de caminar en la
adolescencia
- CMTX
- dominante
- ligada al cromosoma X
- ligada al cromosoma X
- dominante
- Tipo 1 (CMT1) o desmielinizante
- HEREDITARIA NEURODEGENERATIVA
- LEUCODISTROFIA
- desórdenes genéticos hereditarios resultante de la degeneración de la grasa de
la vaina de mielina que cubre las fibras nerviosas del cerebro, y las glándulas
adrenales los nervios espinales
- SÍNTOMAS
- Alteraciones motoras. • Las crisis convulsivas son raras y
el deterioro cognitivo no es precoz. • Lactante: predomina
la detención e involución del desarrollo psicomotor,
irritabilidad, dificultad de alimentación .
- • Parálisis espásticas progresivas • Movimientos
anormales • Rigidez de descerebración • Muerte. •
Adulto: predominan los trastornos psiquiátricos
- Alteraciones motoras. • Las crisis convulsivas son raras y
el deterioro cognitivo no es precoz. • Lactante: predomina
la detención e involución del desarrollo psicomotor,
irritabilidad, dificultad de alimentación .
- edad
- Desde el nacimiento
hasta la edad adulta
- Desde el nacimiento
hasta la edad adulta
- tipos
- Con defecto metabólico conocido
- Con defecto metabólico desconocido
- Encefalopatías mitocondriales
- Distrofia muscular congénita
- Con defecto metabólico conocido
- desórdenes genéticos hereditarios resultante de la degeneración de la grasa de
la vaina de mielina que cubre las fibras nerviosas del cerebro, y las glándulas
adrenales los nervios espinales
- AUTOSOMICA RECESIVA
- LEUCODISTROFIA
- Charcot-Marie-Tooth
atrofia muscular peroneal
- ADQUIRIDAS
- SINDROME DE GUILLAN BARRE
- polirradiculoneuropatía inflamatoria
aguda de carácter progresivo
- por
- inflamación de los nervios
periféricos
- secundario a
- factores autoinmunes
- el daño a la mielina se produce en un periodo de tres semanas
- el daño a la mielina se produce en un periodo de tres semanas
- factores autoinmunes
- secundario a
- inflamación de los nervios
periféricos
- por
- etiopatogenia
- Campylobacter jejuni, Cytomegalovirus y
virus de Epstein Barr, también detectó
infecciones por Mycoplasma pneumoniae,
virus de la hepatitis, herpes simple,
mononucleosis infecciosa y SIDA (HIV).
También se ha asociado con vacunación
(influenza, antirrábica
- Campylobacter jejuni, Cytomegalovirus y
virus de Epstein Barr, también detectó
infecciones por Mycoplasma pneumoniae,
virus de la hepatitis, herpes simple,
mononucleosis infecciosa y SIDA (HIV).
También se ha asociado con vacunación
(influenza, antirrábica
- sintomas
- Los síntomas típicos son: a) Debilidad muscular
o pérdida de la función muscular (parálisis)
paralisis flacida 1. la debilidad comienza en los
pies y las piernas y puede progresar hacia arriba
hasta los brazos y la cabeza 2. puede empeorar
rápidamente entre 24 y 72 horas 3. puede
comenzar en los brazos y progresar hacia abajo
4. puede ocurrir en los brazos y las piernas al
mismo tiempo 5. puede ocurrir únicamente en
los nervios craneanos 6. en los casos leves, es
posible que no ocurra ni la parálisis ni la
debilidad
- falta de coordinación
- Cambios en la sensibilidad
- Sensibilidad o dolor muscular (puede ser
similar al dolor por calambres)
- Dificultad respiratoria
- Los síntomas típicos son: a) Debilidad muscular
o pérdida de la función muscular (parálisis)
paralisis flacida 1. la debilidad comienza en los
pies y las piernas y puede progresar hacia arriba
hasta los brazos y la cabeza 2. puede empeorar
rápidamente entre 24 y 72 horas 3. puede
comenzar en los brazos y progresar hacia abajo
4. puede ocurrir en los brazos y las piernas al
mismo tiempo 5. puede ocurrir únicamente en
los nervios craneanos 6. en los casos leves, es
posible que no ocurra ni la parálisis ni la
debilidad
- diagnóstico
- Debilidad progresiva
en varias extremidades
• Arreflexia
- Progresión desde unos días a 4 semanas •
Relativa simetría • Alteraciones
sensoriales leves • Compromiso de pares
craneales incluyendo el facial •
Recuperación que comienza 2 a 4
semanas después de detenerse la
progresión • Disfunción autonómica •
Ausencia de fiebre una vez instalado el
síndrome
- Estudio del líquido
cefalorraquídeo • Proteínas
elevadas después de una semana
• Menos de 10 linfocitos /mm3
- Pruebas electrofisiológicas • Conducción nerviosa
lenta • Latencias distales prolongadas • Respuestas
tardías anormales
- Velocidad de conducción nerviosa
enlentecida. 2. Bloqueo parcial de la
conducción motora. 3. Dispersión
temporal anormal. 4. Latencias distales
prolongadas.
- Velocidad de conducción nerviosa
enlentecida. 2. Bloqueo parcial de la
conducción motora. 3. Dispersión
temporal anormal. 4. Latencias distales
prolongadas.
- clasificación severidad
- Debilidad progresiva
en varias extremidades
• Arreflexia
- Tratamiento
- Plasmaféresis
- se extrae completamente la sangre del
cuerpo y se procesa de forma que los
glóbulos blancos, glóbulos rojos y
plaquetas se separen del plasma. Las
células de la sangre se devuelven luego
al paciente sin el plasma, el cual el
organismo sustituye rápidamente.
- se extrae completamente la sangre del
cuerpo y se procesa de forma que los
glóbulos blancos, glóbulos rojos y
plaquetas se separen del plasma. Las
células de la sangre se devuelven luego
al paciente sin el plasma, el cual el
organismo sustituye rápidamente.
- Inmunoglobulina G humana IV
- Rehabilitación
- TTo FT
https://www.youtube.com/watch?v=1VY4Mv3ftkw
- movilidad
articular
- cambios de
decubito
- ejercicios pasivos activos asistidos y activos resistidos
- alineación postural
- evitar retracciones
- deformidades
- deformidades
- MaNTENER EL
CONTROL MOTOR,
COORDINACIÓN Y
ESQUEMA CORPORAL
- Bobath, Perfetti Kabath
- Reeducación sensorial
- pilates
- RPG
- rhb marcha
- movilidad
articular
- TTo FT
https://www.youtube.com/watch?v=1VY4Mv3ftkw
- Plasmaféresis
- Variantes del SGB.
- Neuropatía axonal sensitivo motora aguda
- Es una variedad asociada a mal
pronóstico, con inicio de la VM antes
de los 7 dias con tetraparesia profunda
y VM prolongada.
- estudios histopatológicos demuestran
desmielinización mediada por macrófagos e
infiltrados de linfocitos T
- muestra una recuperación más lenta que el SGB
clásico, y las secuelas motoras y sensitivas son
frecuentes
- muestra una recuperación más lenta que el SGB
clásico, y las secuelas motoras y sensitivas son
frecuentes
- estudios histopatológicos demuestran
desmielinización mediada por macrófagos e
infiltrados de linfocitos T
- Es una variedad asociada a mal
pronóstico, con inicio de la VM antes
de los 7 dias con tetraparesia profunda
y VM prolongada.
- Neuropatía axonal
motora aguda (AMAN)
- Se asocia con mayor frecuencia a
infección por Campylobacter jejuni
- cuadro clínico no es necesariamente
grave y la gravedad depende de la
extensión de la lesión axonal.
- casos con exclusivo compromiso distal la
recuperación es rápida y completa
- casos con exclusivo compromiso distal la
recuperación es rápida y completa
- cuadro clínico no es necesariamente
grave y la gravedad depende de la
extensión de la lesión axonal.
- Se asocia con mayor frecuencia a
infección por Campylobacter jejuni
- Síndrome de Miller Fisher (SMF)
- se caracteriza por la asociación de ataxia, oftalmoplejía y arreflexia
- evolución del SMF es favorable y no se acompaña de insuficiencia respiratoria
ni de paresia severa de extremidades
- se caracteriza por la asociación de ataxia, oftalmoplejía y arreflexia
- Polineuropatía desmielinizante inflamatoria crónica (CIDP)
- se diferencia por la progresión de los signos y síntomas durante un
período superior a los 28 días, o rápida progresión seguida de recidivas
repetidas del cuadro
- los pacientes demoran más de 2 meses en desarrollar el cuadro
completo
- recuperación es total en un período de meses,
- los pacientes demoran más de 2 meses en desarrollar el cuadro
completo
- se diferencia por la progresión de los signos y síntomas durante un
período superior a los 28 días, o rápida progresión seguida de recidivas
repetidas del cuadro
- Neuropatía sensitivo motora axonal
aguda (NSMAA)
- Se observa lesión severa de los axones sensitivos y
motores con escaso infiltrado linfocitario, sin
desmielinización; los cambios se extienden a las
porciones proximales de las raíces nerviosas; se
relaciona con inicio fulminante y déficits
sensitivos.
- Se observa lesión severa de los axones sensitivos y
motores con escaso infiltrado linfocitario, sin
desmielinización; los cambios se extienden a las
porciones proximales de las raíces nerviosas; se
relaciona con inicio fulminante y déficits
sensitivos.
- Polineuropatía sensitivo motora
desmielinizante aguda.
- Es la más frecuente en países desarrollados (90%).
Los estudios histopatológicos demuestran
desmielinización mediada por macrófagos e
infiltrados de linfocitos T.
- Es la más frecuente en países desarrollados (90%).
Los estudios histopatológicos demuestran
desmielinización mediada por macrófagos e
infiltrados de linfocitos T.
- Neuropatía axonal sensitivo motora aguda
- fisiopatologia
- se produce infiltración por linfocitos
T. Posteriormente ocurre
desmielinización en axones sensitivos
y motores, mediada por macrófagos,
que penetran la membrana basal de
las células de Schwann, desgarran las
laminillas de mielina dejando a los
axones expuestos, con degeneración
de fibras, raíces, nervios proximales y
distales
- se produce infiltración por linfocitos
T. Posteriormente ocurre
desmielinización en axones sensitivos
y motores, mediada por macrófagos,
que penetran la membrana basal de
las células de Schwann, desgarran las
laminillas de mielina dejando a los
axones expuestos, con degeneración
de fibras, raíces, nervios proximales y
distales
- polirradiculoneuropatía inflamatoria
aguda de carácter progresivo
- metabólicas
- neuropatía diabética
- sensitivo-motora
- exposición prolongada a
niveles elevados de glucosa
causa daño de los nervios
- autónoma
- causa cambios en funciones digestivas, intestinales y vesicales, en la respuesta sexual y en la
transpiración. También puede afectar los nervios asociados con el corazón y aquellos que controlan
la presión arterial, así como los nervios en los pulmones y los ojos
- causa cambios en funciones digestivas, intestinales y vesicales, en la respuesta sexual y en la
transpiración. También puede afectar los nervios asociados con el corazón y aquellos que controlan
la presión arterial, así como los nervios en los pulmones y los ojos
- periférica
- causa dolor o pérdida de
sensación en los dedos del
pie, en los pies, las piernas, las
manos y los brazos.
- causa dolor o pérdida de
sensación en los dedos del
pie, en los pies, las piernas, las
manos y los brazos.
- proximal
- dolor en los muslos, caderas o nalgas
y produce debilidad en las piernas.
- dolor en los muslos, caderas o nalgas
y produce debilidad en las piernas.
- focal
- debilitamiento repentino
de un nervio o un grupo
de nervios, causando
debilidad muscular o
dolor. Cualquier nervio
en el cuerpo puede verse
afectado.
- debilitamiento repentino
de un nervio o un grupo
de nervios, causando
debilidad muscular o
dolor. Cualquier nervio
en el cuerpo puede verse
afectado.
- autónoma
- exposición prolongada a
niveles elevados de glucosa
causa daño de los nervios
- sensitivo-motora
- neuropatía diabética
- alcholicas
- consumo excesivo de alcohol durante largos períodos de tiempo.
- aumentar las toxinas dentro del cuerpo de una persona como
etanol y acetaldehído
- malos hábitos de vida y nutricionales
- síntomas
- pérdida de la sensibilidad hormigueo en las
pies/manos tobillos débiles debilitamiento de
los músculos y una sensación de ardor en los
pies
- pérdida de la sensibilidad hormigueo en las
pies/manos tobillos débiles debilitamiento de
los músculos y una sensación de ardor en los
pies
- síntomas
- malos hábitos de vida y nutricionales
- aumentar las toxinas dentro del cuerpo de una persona como
etanol y acetaldehído
- consumo excesivo de alcohol durante largos períodos de tiempo.
- Neuropatía e insuficiencia
renal
- neuropatía sensitivo-motora simétrica, de
predominio distal y siempre más manifiesta en
las extremidades inferiores
- secundaria a la acumulación de metabolitos o toxinas
dializables que habitualmente excreta el riñón, sospecha
que se ve reforzada por la similitud entre la polineuropatía
urémica y muchas de las tóxicas
- disestesias quemantes, los calambres
musculares, y el síndrome de piernas
inquieta
- ausencia de reflejos aquíleos, y la alteración
de la sensibilidad vibratoria, artrocinética y
táctil.
- La debilidad y atrofia
son tardías
- La debilidad y atrofia
son tardías
- ausencia de reflejos aquíleos, y la alteración
de la sensibilidad vibratoria, artrocinética y
táctil.
- disestesias quemantes, los calambres
musculares, y el síndrome de piernas
inquieta
- secundaria a la acumulación de metabolitos o toxinas
dializables que habitualmente excreta el riñón, sospecha
que se ve reforzada por la similitud entre la polineuropatía
urémica y muchas de las tóxicas
- axonopatía distal, con atrofia axonal, pérdida
discreta de fibras y degeneración secundaria de la
mielina
- neuropatía sensitivo-motora simétrica, de
predominio distal y siempre más manifiesta en
las extremidades inferiores
- SINDROME DE GUILLAN BARRE
- Neuropatías asociadas a
agentes infecciosos
- Virus de la inmunodeficiencia
humana
- desmielinización segmentaria
- forma más común
- polineuritis sensitiva simétrica y
distal,
- disestesias
dolorosas
- degeneración axonal que afecta
predominantemente a las fibras
de gran tamaño
- disestesias
dolorosas
- polineuritis sensitiva simétrica y
distal,
- forma más común
- desmielinización segmentaria
- Lepra
- causada
- Mycobacterium leprae
(bacilo de Hansen).
- síntoma
- pérdida de la sensibilidad,
especialmente la termoalgé-
sica
- nervios se hipertrofian
pudiéndose palpar y
haciéndose
especialmente
vulnerables a
compresiones y
traumatismos
- nervios se hipertrofian
pudiéndose palpar y
haciéndose
especialmente
vulnerables a
compresiones y
traumatismos
- pérdida de la sensibilidad,
especialmente la termoalgé-
sica
- síntoma
- Mycobacterium leprae
(bacilo de Hansen).
- causada
- Virus de la inmunodeficiencia
humana
- Neuropatía del enfermo grave
- enfermos intubados y ventilados que
han pasado por una fase de disfunción
multiorgánica
- dificultad para retirar
el ventilador
- paresia flácida de las
extremidades y depresión de
los reflejos osteotendinosos
- electroneurografía sugiere una
degeneración axonal primaria, lo que
permite diferenciar el cuadro de un
Guillain-Barré
- electroneurografía sugiere una
degeneración axonal primaria, lo que
permite diferenciar el cuadro de un
Guillain-Barré
- paresia flácida de las
extremidades y depresión de
los reflejos osteotendinosos
- dificultad para retirar
el ventilador
- enfermos intubados y ventilados que
han pasado por una fase de disfunción
multiorgánica
- https://www.youtube.com/watch?v=h4ZJiwL6IEM
Recursos multimedia adjuntos
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
¿Quieres crear tus propios Mapas Mentales gratis con GoConqr? Más información.