5405099
Description
Flashcards by cryptic_arcana, updated more than 1 year ago
|
|
Created by cryptic_arcana
over 8 years ago
|
|
| Question | Answer |
| Cellular response to stress and noxious stimuli | |
| Adaptations to stress | Can be physiological or pathological Hypertrophy - increase in cell size Hyperplasia - increase in cell number Atrophy - decrease in cell size Metaplasia - reversible change in cell type |
| Hypertrophy | Increase in cell size, resulting in increased organ size. Caused either by increased functional demand (e.g. muscular hypertrophy in exercise) or stimulation by growth factors or other hormones (e.g. uterine smooth muscle hypertrophy in pregnancy). |
| Hyperplasia | Increase in cell number, occuring in tissues capable of replication. Physiological: Hormonal - induced by growth factors or hormones Compensatory - growth of residual tissue after loss or resection of part of an organ (e.g. liver) Pathological: Usually hormone- or growth factor-mediated (e.g. fibroblast hyperplasia induced by leukocyte-produced growth factors in injury) Disappears if the hormonal signal inducing hyperplasia is removed. |
| Atrophy | Due to decreased workload, loss of innervation, reduced blood supply, inadequate nutrition or loss of endocrine stimulation. Represents a decrease in cell size to a form with lower energy requirements but at which cell survival is still possible. Combination of decreased protein synthesis (due to decreased metabolic activity) and increased protein degradation. Protein degradation occurs mainly via the ubiquitin-proteasome pathway. Nutrient deficiency and disuse activate ubiquitin ligases, which attach ubiquitin to cellular proteins, which are then targeted by proteasomes. Also often accompanied by autophagy. |
| Metaplasia | A reversible process whereby a cell sensitive to a particular stress is replaced by another cell type less sensitive to the stressor, e.g. columnar epithelial metaplasia in the distal oesophagus from gastrooesophageal reflux. Although the new cell type is better able to withstand the environment, important functions and protective features of the previous cell type are lost. Usually induced by altered differentiation pathway of tissue stem cells. Stressors which cause metaplastic change may, if persistent, predispose towards further neoplastic change. |
| Causes of cell injury | - Hypoxia - Toxins - Infectious agents - Immune reactions - Genetic factors - Nutritional imbalances - both malnutrition and excess nutrition (e.g. obesity) - Physical trauma - mechanical, thermal, electrical - Ageing |
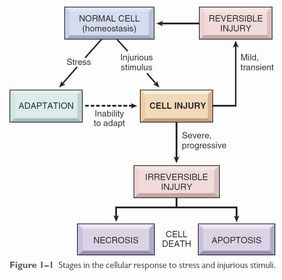
| Reversible cellular injury | Injury occurs when cells are stressed so severely that they can no longer adapt, or are exposed to inherently damaging agents (e.g. extreme heat) or suffer from intrinsic abnormalities. In early cell injury, the changes are reversible if the damaging stimulus is removed. At this stage, there are significant structural changes - swelling of ER and mitochondria, cell membrane blebs - but changes have not progressed to nuclear disruption or severe membrane damage. |
| Two types of reversible cellular injury | Cellular swelling - due to failure of energy-dependent ion pumps, leading to an inability to maintain ionic and fluid homeostasis and osmotic swelling of the cell. Fatty change - due to hypoxic injury and some forms of metabolic and toxic injury. Forms lipid vacuoles within the cell. |
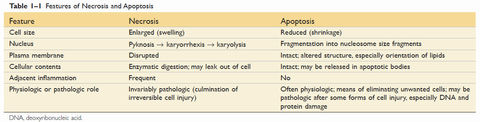
| Cell death | Apoptosis: - Nuclear dissolution without complete membrane disruption - Can be physiological as well as pathological - Does not elicit an inflammatory response Necrosis: - Membrane damage causes enzyme leakage from lysosomes, which then digest the cell. - Inflammatory response due to products of cell breakdown and intracellular contents leaking into extracellular space through the damaged cell membrane. - Always pathological |
| Apoptosis vs necrosis | |
| Apoptosis - physiological causes | - Programmed destruction of cells during embryogenesis and normal development - Breakdown of hormone-dependent tissues when deprived of hormone - Cell loss in proliferating cells (e.g. intestinal epithelium) - Elimination of cells which have served their useful purpose - Elimination of self-reactive lymphocytes - Cell death induced by cytotoxic T-lymphocytes, as a defence against viruses and neoplastic cells |
| Apoptosis - pathological causes | - DNA damage - if repair mechanisms cannot repair the damage, apoptosis is triggered as a defence mechanism against neoplasia and mutation. - Accumulation of misfolded proteins - excessive accumulation in the ER leads to ER stress and apoptosis - Cell death in certain infections, particularly viral - may be triggered by the virus itself, or by T-lymphocytes as a defence mechanism - Pathological atrophy |
| Apoptosis - mechanisms | - Results from activation of caspases (cysteine proteases - Depends on balance between production of pro-apoptotic and anti-apoptotic proteins - Activation of initiator enzymes (caspase-9 and -8) leads to the cleavage of other pro-enzymes into active forms in a cascade which ultimately leads to the activation of nucleases to degrade DNA and nuclear proteins, and other enzymes to degrade the cytoskeleton and nuclear matrix, leading to fragmentation of the cell into apoptotic bodies - Apoptotic bodies express ligands for phagoytic cell receptors (phosphatidylserine), leading to their disposal by phagocytes Two pathways to activation - intrinsic (mitochondrial) and extrinsic (death receptor) |
| Apoptosis - intrinsic (mitochondrial) pathway | - Mitochondria contain several pro-apoptotic proteins (e.g. cytochrome C) that neutralise endogenous anti-apoptotic proteins - Apoptosis via the intrinsic pathway is determined by the permeability of mitochondria, which is controlled by the Bcl-2 family of proteins - Increased permeability allows the leakage of pro-apoptotic proteins into the cytoplasm. - Cytochrome C activates caspase-9, the initiator caspase for the intrinsic pathway - Intrinsic pathway is responsible for apoptosis in most situations |
| Apoptosis - extrinsic (death receptor) pathway | - Most cells have 'death receptors' on the surface - Usually part of the TNF receptor family, in particular the Type 1 TNF receptor and the FAS receptor - Death receptors contain a 'death domain' in the cytoplasmic region of the receptor - When the receptor is activated, the death domain binds adapter proteins, which then activates caspase-8, the initiator of the extrinsic pathway - Caspase-8 also cleaves and activates a pro-apoptotic protein in the Bcl-2 family, feeding into the intrinsic pathway - Extrinsic pathway is involved in the elimination of self-reactive lymphocytes and the killing of target cells by CD-8 T-lymphocytes |
| Necrosis - morphology | Coagulative - e.g. infarcts (except brain). Underlying tissue structure is maintained - enzymes are likely denatured, inhibiting proteolysis. Liquefactive - formation of pus Gangrenous Caseous - associated with mycobacterial infection. Tissue architecture is completely destroyed. Fat necrosis - from activation of pancreatic lipases Fibrinoid necrosis - immune-mediated. Deposition of immune complexes. |
| Necrosis - mechanisms | - Depletion of ATP - Mitochondrial damage - Influx of calcium - Oxidative stress - Increased membrane permeability |
| Necrosis - depletion of ATP | - Usually due to hypoxia, reduced nutrient supply, mitochondrial damage and some toxins (e.g. cyanide) - Reduced activity of Na-K ATPase causes influx of sodium and efflux of potassium - Osmotic effect causes influx of water, causing swelling of the cell and endoplasmic reticulum - Compensatory increase in anaerobic glycolysis rapidly depletes glycogen stores and produces lactic acid, decreasing the pH and reducing the activity of many cellular enzymes - Failure of ATP-dependent Ca pumps leads to influx of calcium |
| Necrosis - mitochondrial damage | - Mitochondria are susceptible to many forms of damage, including hypoxia, toxins and radiation Several effects of mitochondrial damage: - Reduced ATP production, leading to ATP depletion - Abnormal oxidative phosphylation leads to the formation of reactive oxygen species and increased oxidative stress - Increased mitochondrial permeability causes loss of membrane potential and pH changes, further inhibiting mitochrondrial function - Increased permeability and/or destruction of mitochondria also release proteins (chiefly cytochrome C) that feed into the intrinsic pathway of apoptosis |
| Necrosis - oxidative stress | - Increased production of reactive oxygen species can result from ionising radiation, metabolism of certain chemicals/toxins and inflammation, where ROS are produced by leukocytes - Chiefly peroxide (via superoxide) and nitric oxide ROS cause damage through three main mechanisms: - Peroxidation of lipid membranes - Cross-linking and other oxidation of proteins - DNA damage Protective mechanisms: - Enzymes (superoxide dismutases, glutathione peroxidases and catalases) which break down peroxide - Antioxidants (e.g. Vitamin A, C and E) block the formation of ROS or scavenge them once formed |
{kind=link}
{kind=link}
Want to create your own Flashcards for free with GoConqr? Learn more.