14381503
Description
Mind Map by Ender Arlid, updated more than 1 year ago
|
|
Created by Ender Arlid
over 6 years ago
|
|
Enfermedades Cromosómicas
- Estas enfermedades se deben a alteraciones en la estructura de los cromosomas, como pérdida o deleción cromosómica
- encontramos las siguientes:
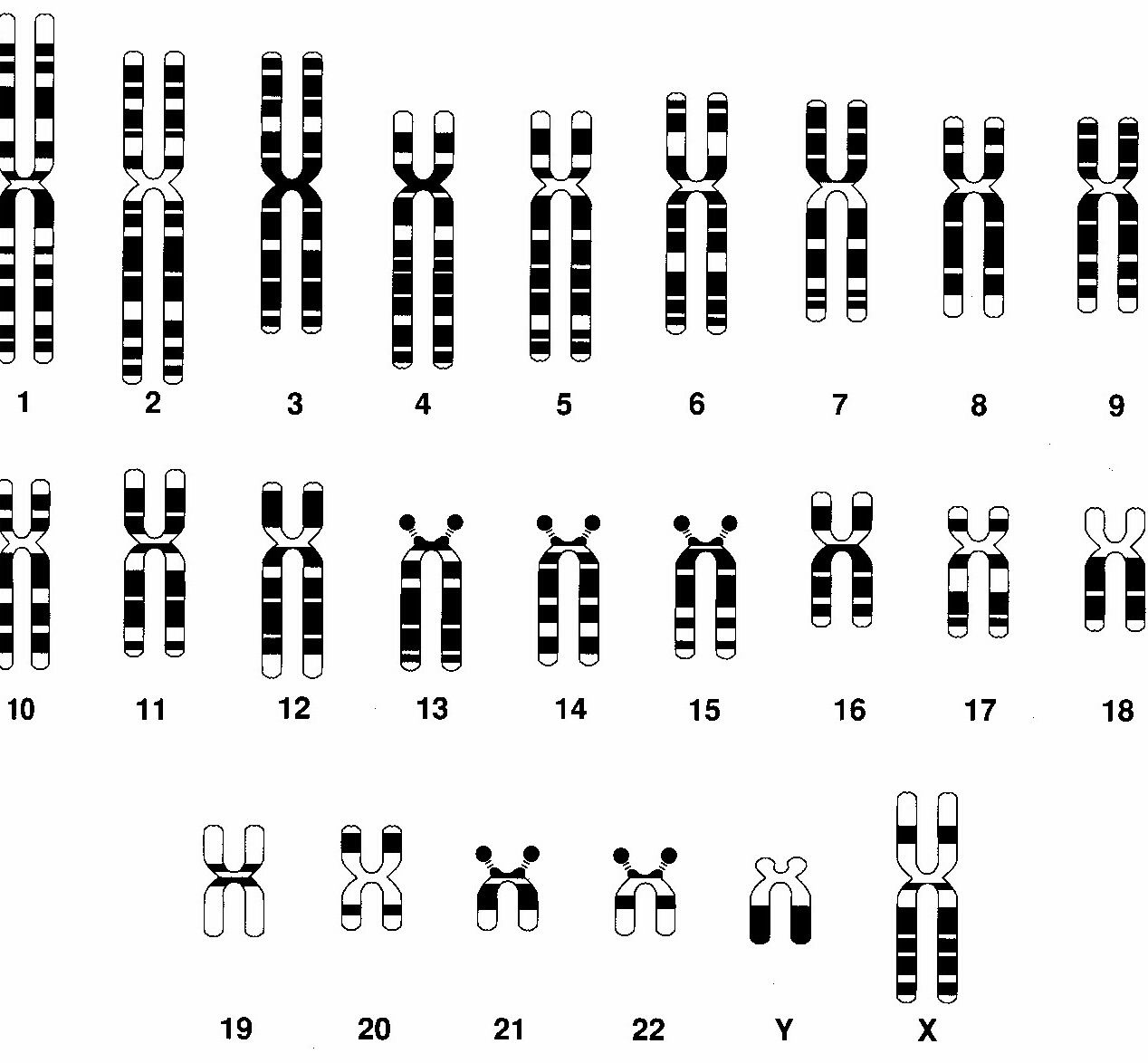
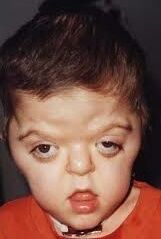
- Síndrome de Down o trisomía 21: debida a la presencia de una copia adicional de la zona crítica en 21q22.1, que puede deberse a una trisomía 21 en más del 94% de los casos, una translocación con otros cromosomas (heredado o no) en el 3.3%, la coexistencia de una línea celular con la trisomía 21, otras, lo que se denomina mosaicismo en el 2.4%. Se asocia a una edad materna avanzada. Presentan hipotonía, talla baja, braquicefalia, facie aplanada, epicanto, orejas pequeñas con hélix plegado y protrusión lingual. Tienen exceso de piel en la nuca, cardiopatía congénita en el 50%, malformaciones gastrointestinales, pueden hacer reacciones leucemoides y neoplasias hematológicas.
- Síndrome de Edwards o trisomía 18: debida a la presencia de tres cromosomas 18. También hay una correlación con edad materna avanzada. El pronóstico de sobrevida es muy pobre.
- Síndrome de Patau o trisomía 13: debida a la presencia de tres cromosomas 13. Se relaciona con edad materna avanzada, es clínicamente muy severa y generalmente letal antes de los tres meses de edad. El fenotipo incluye malformaciones del sistema nervioso central, hipotelorismo ocular, microftalmia, fisura labiopalatina,
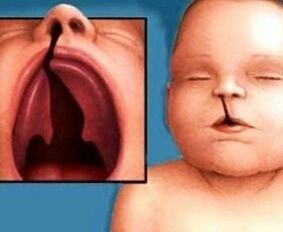
- Síndrome de Wolf Hirschhorn o 4p-: debida a la deleción parcial de un segmento del brazo corto del cromosoma 4 que involucre 4p16.3. Presentan retraso severo de crecimiento con microcefalia. Tienen un perfil característico con el puente nasal alto, nariz ganchuda, filtrum corto, micrognatia, comisuras orales hacia abajo, fisura labiopalatina y orejas de implantación baja.
- Síndrome de Cri du Chat o 5p-: debida a la deleción parcial de un segmento del brazo corto del cromosoma 5 que involucre 5p15.2. Aunque la mayoría de las deleciones ocurren como mutaciones de novo, en alrededor de un 12% resultan de una translocación presente en uno de los padres. Microcefalia, fascie redonda, hipertelorismo ocular, epicanto, estrabismo, fisuras palpebrales hacia abajo, micrognatia, orejas de implantación baja, cardiopatía congénita, e hipotonía, con severo retardo psicomotor.
- Síndrome de Turner o monosomía X: presencia de un sólo cromosoma X, también pueden verse diversas alteraciones estructurales en un cromosoma X y no es rara la presencia de dos o más líneas celulares diferentes. Talla baja, disgenesia gonadal, cuello alado, linfedema del dorso de pies y manos, uñas angostas, cúbito valgo, paladar ojival, tórax ancho, malformaciones renales y cardiovasculares.
- Síndrome Klinefelter o 47,XXY: presencia de un cromosoma X adicional en un varón. Los rasgos clínicos en niños son mínimos y el diagnóstico puede retrasarse hasta la adolescencia, por talla alta, trastornos de aprendizaje y testículos pequeños. Infertilidad.
- Síndrome de Down o trisomía 21: debida a la presencia de una copia adicional de la zona crítica en 21q22.1, que puede deberse a una trisomía 21 en más del 94% de los casos, una translocación con otros cromosomas (heredado o no) en el 3.3%, la coexistencia de una línea celular con la trisomía 21, otras, lo que se denomina mosaicismo en el 2.4%. Se asocia a una edad materna avanzada. Presentan hipotonía, talla baja, braquicefalia, facie aplanada, epicanto, orejas pequeñas con hélix plegado y protrusión lingual. Tienen exceso de piel en la nuca, cardiopatía congénita en el 50%, malformaciones gastrointestinales, pueden hacer reacciones leucemoides y neoplasias hematológicas.
- https://m.youtube.com/watch?v=_yLOCATc2GI
- Noticia
- Prueban una nueva terapia génica para tratar la enfermedad de Hunter
- Que es la enfermedad de Hunter ?
- La enfermedad de Hunter es una enfermedad rara que afecta principalmente a niños causando graves daños a diversos órganos. Hasta ahora era incurable, pero una nueva terapia génica abre esperanzas a su tratamiento.
- La enfermedad de Hunter es una enfermedad rara que afecta principalmente a niños causando graves daños a diversos órganos. Hasta ahora era incurable, pero una nueva terapia génica abre esperanzas a su tratamiento.
- El nuevo tratamiento, que ya está en la fase de ensayos clínicos en pacientes, consiste en reemplazar el gen que causa la enfermedad de Hunter en la médula ósea de los pequeños afectados.Pero, además, se añade una clase de lípidos o grasas, llamada triacilglicéridos (TAG) a la enzima IDS para permitir que llegue al cerebro y tenga más efectividad.
- https://www.webconsultas.com/noticias/bebes-y-ninos/prueban-una-nueva-terapia-genica-para-tratar-la-enfermedad-de-hunter
- https://www.webconsultas.com/noticias/bebes-y-ninos/prueban-una-nueva-terapia-genica-para-tratar-la-enfermedad-de-hunter
- Que es la enfermedad de Hunter ?
- Prueban una nueva terapia génica para tratar la enfermedad de Hunter
Media attachments
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Want to create your own Mind Maps for free with GoConqr? Learn more.