16255873
Description
Mind Map by Alejandra Castelo, updated more than 1 year ago
|
|
Created by Alejandra Castelo
about 6 years ago
|
|
Malformaciones congénitas
- Anormalidades del
desarrollo
- Son cuadros congénitos en que son atípicas las características del desarrollo cromosómico, gonadal o
anatómico sexual
- Son cuadros congénitos en que son atípicas las características del desarrollo cromosómico, gonadal o
anatómico sexual
- Seudohermafroditismo
femenino
- La discrepancia entre el sexo gonadal (46,XX) y la imagen fenotípica de los genitales externos
(masculinizados) es consecuencia de la exposición excesiva del feto a andrógenos.
- La discrepancia entre el sexo gonadal (46,XX) y la imagen fenotípica de los genitales externos
(masculinizados) es consecuencia de la exposición excesiva del feto a andrógenos.
- Disgenesia
gonadal
- Es el desarrollo anormal de las gónadas, suele ser consecuencia de la falta de disyunción de cromosomas de
ambos padres, y da origen a estrías gonadales. La mayoría curso con un cariotipo 45 X, o síndrome de Turner,
que se acompaña de cúbito valgo, anormalidades cardiacas (coartación aórtica principalmente), anomalías
renales, deficiencias auditivas, otitis media, mastoiditis, hipertensión, aclorhidria, diabetes mellitus y
tiroiditis de Hashimoto. En la adolescnecia se manifiesta por talla corta, genitales femeninos prepuberales y
amenorrea primaria
- Es el desarrollo anormal de las gónadas, suele ser consecuencia de la falta de disyunción de cromosomas de
ambos padres, y da origen a estrías gonadales. La mayoría curso con un cariotipo 45 X, o síndrome de Turner,
que se acompaña de cúbito valgo, anormalidades cardiacas (coartación aórtica principalmente), anomalías
renales, deficiencias auditivas, otitis media, mastoiditis, hipertensión, aclorhidria, diabetes mellitus y
tiroiditis de Hashimoto. En la adolescnecia se manifiesta por talla corta, genitales femeninos prepuberales y
amenorrea primaria
- Hermafroditismo verdadero
- Las personas afectadas tienen tejido gonadal ovárico y testicular. El cariotipo más común en los
hermafroditas verdaderos es 46,XX. El fenotipo del hermafrodita verdadero 46,XX incluye un ovotestículo
unilateral con un ovario o un testículo en el lado contrario, o en ambos lados, ovotestículo. El sitio en que
están las gónadas varía desde el abdomen a la ingle o el escroto.
- Las personas afectadas tienen tejido gonadal ovárico y testicular. El cariotipo más común en los
hermafroditas verdaderos es 46,XX. El fenotipo del hermafrodita verdadero 46,XX incluye un ovotestículo
unilateral con un ovario o un testículo en el lado contrario, o en ambos lados, ovotestículo. El sitio en que
están las gónadas varía desde el abdomen a la ingle o el escroto.
- Síndrome de
Klinefelter
- El síndrome de Klinefelter (47,XXY) afecta a 1 de cada 500 neonatos vivos o 1 a 2% de todos los varones.
Las personas con este trastorno tienden a ser de talla corta, varones subvirilizados con ginecomastia y con
testículos firmes y pequeños. Su fecundidad disminuye enormemente por el hipogonadismo y están
expuestos a un mayor riesgo de tumores de células germinativas, osteoporosis y cáncer de mama.
- El síndrome de Klinefelter (47,XXY) afecta a 1 de cada 500 neonatos vivos o 1 a 2% de todos los varones.
Las personas con este trastorno tienden a ser de talla corta, varones subvirilizados con ginecomastia y con
testículos firmes y pequeños. Su fecundidad disminuye enormemente por el hipogonadismo y están
expuestos a un mayor riesgo de tumores de células germinativas, osteoporosis y cáncer de mama.
- Extrofia
vesical
- La extrofia vesical es consecuencia de que la membrana cloacal no fue “reforzada” por la invaginación del
mesodermo. Los signos acompañantes suelen incluir genitales externos anormales y sínfisis del pubis
ensanchada, causada por la rotación de los iliacos hacia afuera.
- La extrofia vesical es consecuencia de que la membrana cloacal no fue “reforzada” por la invaginación del
mesodermo. Los signos acompañantes suelen incluir genitales externos anormales y sínfisis del pubis
ensanchada, causada por la rotación de los iliacos hacia afuera.
- Defectos del
clítoris
- duplicación de dicho órgano, uretra fálica femenina o
clitoromegalia.
- duplicación de dicho órgano, uretra fálica femenina o
clitoromegalia.
- Anomalías de
vulva
- Totales
- Ausencia de vulva. Forma parte de una importante anomalía en la morfogénesis del polo caudal embrionario,
con soldadura de miembros inferiores. El feto no es viable. * Duplicidad de la vulva. Muy rara, se asocia a
otras malformaciones, a veces mortales para el feto. * Hipoplasia. La vulva es muy estrecha y los labios
mayores tienen dimensiones infantiles. Puede dificultar las relaciones sexuales, requiriendo una apertura
quirúrgica, e incluso puede interferir el momento del parto, preconizándose la cesárea en algunos casos
- Ausencia de vulva. Forma parte de una importante anomalía en la morfogénesis del polo caudal embrionario,
con soldadura de miembros inferiores. El feto no es viable. * Duplicidad de la vulva. Muy rara, se asocia a
otras malformaciones, a veces mortales para el feto. * Hipoplasia. La vulva es muy estrecha y los labios
mayores tienen dimensiones infantiles. Puede dificultar las relaciones sexuales, requiriendo una apertura
quirúrgica, e incluso puede interferir el momento del parto, preconizándose la cesárea en algunos casos
- Parciales
- Hipertrofia de labios menores * Quistes congénitos. Desaparecen espontáneamente pocas semanas tras el
nacimiento. Si persisten, se debe hacer punción/extirpación.
- Hipertrofia de labios menores * Quistes congénitos. Desaparecen espontáneamente pocas semanas tras el
nacimiento. Si persisten, se debe hacer punción/extirpación.
- Totales
- Anomalías de mama
- Alteraciones de
número
- Amastia y atelia. Son muy raras y en general integradas en un síndrome malformativo regional. * Polimastia y
politelia. Las mamas y pezones ectópicos y supernumerarios proceden de la persistencia anormal de una
cresta mamaria.
- Amastia y atelia. Son muy raras y en general integradas en un síndrome malformativo regional. * Polimastia y
politelia. Las mamas y pezones ectópicos y supernumerarios proceden de la persistencia anormal de una
cresta mamaria.
- Alteraciones de
tamaño
- Macromastia y
micromastia
- Macromastia y
micromastia
- Alteraciones de
forma
- Ptosis mamaria y pezón umbilicado
- Ptosis mamaria y pezón umbilicado
- Asimetrías
- Alteraciones de
número
- Anomalías
ováricas
- OVario supernumerario, accesorio, ausencia
unilateral
- OVario supernumerario, accesorio, ausencia
unilateral
- Anomalías de los conductos de
Müller
- Casi todos los casos se diagnostican durante una valoración por problemas obstétricos o ginecológicos,
pero en ausencia de síntomas la mayoría no se diagnostican.
- Casi todos los casos se diagnostican durante una valoración por problemas obstétricos o ginecológicos,
pero en ausencia de síntomas la mayoría no se diagnostican.
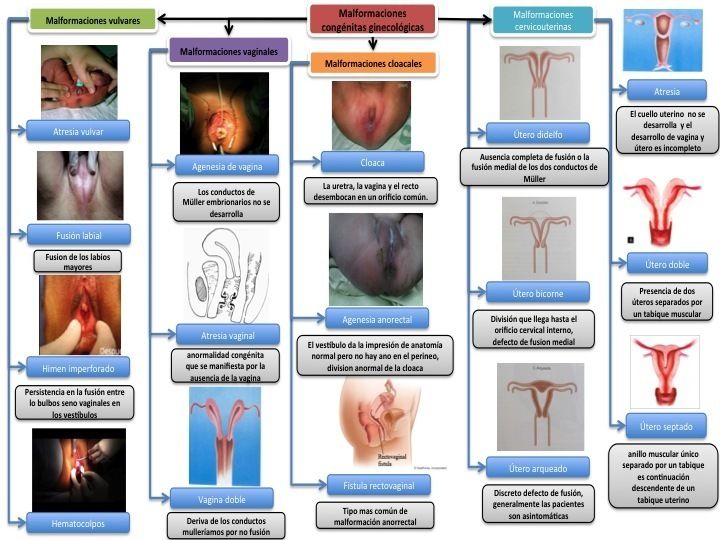
- Útero
- Unicorne
- La maduración esta estancada o defectuosa de un solo conducto de Müller genera un útero unicorne. La
frecuencia de útero unicorne, según la histerosalpingografía en una serie de 1 160 nomalias iterina es el
14%
- La maduración esta estancada o defectuosa de un solo conducto de Müller genera un útero unicorne. La
frecuencia de útero unicorne, según la histerosalpingografía en una serie de 1 160 nomalias iterina es el
14%
- Didelfo
- Se forma cuando no se fusionan los conductos de Müller, esta anomalía se caracteriza por la presencia de
dos cavidades endometriales, cada una de ellas con su cuello uterino. En la mayoría de los casos existe un
tabique vaginal longitudinal que separa ambos cuellos uterinos.
- Se forma cuando no se fusionan los conductos de Müller, esta anomalía se caracteriza por la presencia de
dos cavidades endometriales, cada una de ellas con su cuello uterino. En la mayoría de los casos existe un
tabique vaginal longitudinal que separa ambos cuellos uterinos.
- Bicorne
- La causa del útero bicorne es la fusión lateral incompleta de los conductos de müller. Se caracteriza por la
presencia de dos cavidades endometriales separadas y un solo cuello uterino. Algunas veces la falta de
fusión se extiende hasta el cuello uterino, originando un útero bicorne completo, pero otras es parcial y el
resultado es una anomalía leve.
- La causa del útero bicorne es la fusión lateral incompleta de los conductos de müller. Se caracteriza por la
presencia de dos cavidades endometriales separadas y un solo cuello uterino. Algunas veces la falta de
fusión se extiende hasta el cuello uterino, originando un útero bicorne completo, pero otras es parcial y el
resultado es una anomalía leve.
- Tabicado
- Cuando después de la fusión lateral de los conductor de Müller los segmentos mediales no sufren regresión,
se forma un tabique permanente dentro de la cavidad uterina. Sus contornos varían y dependen de la
cantidad de tejido persistente en la línea media. Algunas veces el tabique se proyecta directamente desde el
fondo uterino y otras veces se extienden hasta el orificio cervicouterino. Otros tabiques son segmentarios,
generando una comunicación parcial del útero dividido. Su estructura histológica va desde fibrosa hasta
fibromuscular.
- Cuando después de la fusión lateral de los conductor de Müller los segmentos mediales no sufren regresión,
se forma un tabique permanente dentro de la cavidad uterina. Sus contornos varían y dependen de la
cantidad de tejido persistente en la línea media. Algunas veces el tabique se proyecta directamente desde el
fondo uterino y otras veces se extienden hasta el orificio cervicouterino. Otros tabiques son segmentarios,
generando una comunicación parcial del útero dividido. Su estructura histológica va desde fibrosa hasta
fibromuscular.
- Arqueado
- Unicorne
Media attachments
{kind=link}
Want to create your own Mind Maps for free with GoConqr? Learn more.