16855852

Enfermedad
poliquística
renal
- ¿qué es?
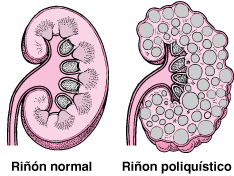
- - Trastorno hereditario
(AD, AR) - Formación
de quístes
(unilaterales o
bilaterales) en riñones
--> aumento de
tamaño de los riñones
--> pérdida de función
- Quistes: sacos
redondos no
cancerosos con
contenido
acuoso
- Quistes: sacos
redondos no
cancerosos con
contenido
acuoso
- - Trastorno hereditario
(AD, AR) - Formación
de quístes
(unilaterales o
bilaterales) en riñones
--> aumento de
tamaño de los riñones
--> pérdida de función
- EPQR
Autosómica
Recesiva
- Etiología
- PKHD1, en el brazo corto del crom. 6 (6p21.2-p12).
Codifica una proteína fibroquistina o poliductina
(FPC). La FPC se encuentra en cilios primarios y el
centrosoma en los túbulos colectores renales y
también en los ductos hepáticos y pancreáticos
- PKHD1, en el brazo corto del crom. 6 (6p21.2-p12).
Codifica una proteína fibroquistina o poliductina
(FPC). La FPC se encuentra en cilios primarios y el
centrosoma en los túbulos colectores renales y
también en los ductos hepáticos y pancreáticos
- Epidemiología
- Trastorno infantil (1er año de
vida), relativamente raro, 1:
6000 a 50,000 nacidos vivos.
Afecta a ambos géneros y
todas las razas por igual.
- Trastorno infantil (1er año de
vida), relativamente raro, 1:
6000 a 50,000 nacidos vivos.
Afecta a ambos géneros y
todas las razas por igual.
- Fisiopatología
- Complicaciones
- FPC forma con la policistina 2 para regular la entrada de
calcio --> cuando hay una pérdida en la FPC --> disminuye
la PC2 --> disminuye señalización de calcio --> no se inhibe
la señal de proliferación --> formación de quistes
- nefropatía intrauterina -->
oligohidramios --> hipoplasia
pulmonar --> morbilidad del
30%
- FPC forma con la policistina 2 para regular la entrada de
calcio --> cuando hay una pérdida en la FPC --> disminuye
la PC2 --> disminuye señalización de calcio --> no se inhibe
la señal de proliferación --> formación de quistes
- Complicaciones
- Manifestaciónes clínicas
- Quistes renales (intrauterinos - adultos) -->
masas abdominales bilaterales, insuficiencia
renal en la infancia. HTA, disfunción tubular
renal : poliuria, enuresis, hiponatremia, acidosis
metabólica hiperclorémica. Infección y rutptura
de quistes --> hematuria (poco común). Fibrosis
hepática --> secundaria a dilatación de
conductos biliares hepáticos --> hipertensión
portal con esplenomegalia. Fibrosis
pancreáticas
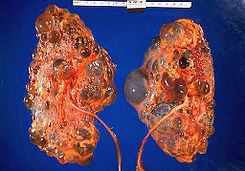
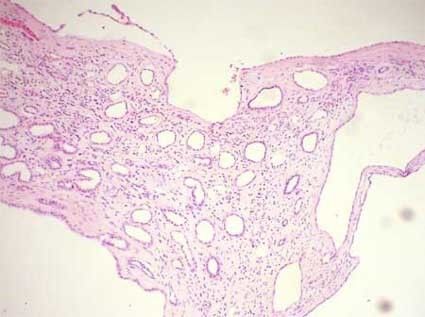
- hitología
- riñones aumentados de
tamaño con dilataciones
quísticas fusiformes y no
obstructivas en los túbulos
colectores, con
adelgazamiento del
parénquima y fibrosis
intersticial
- riñones aumentados de
tamaño con dilataciones
quísticas fusiformes y no
obstructivas en los túbulos
colectores, con
adelgazamiento del
parénquima y fibrosis
intersticial
- Quistes renales (intrauterinos - adultos) -->
masas abdominales bilaterales, insuficiencia
renal en la infancia. HTA, disfunción tubular
renal : poliuria, enuresis, hiponatremia, acidosis
metabólica hiperclorémica. Infección y rutptura
de quistes --> hematuria (poco común). Fibrosis
hepática --> secundaria a dilatación de
conductos biliares hepáticos --> hipertensión
portal con esplenomegalia. Fibrosis
pancreáticas
- Diagnóstico
- Descubrimiento
radiológico de
quistes. Uns
ecografía
abdominal o TC en
riñones
poliquísticos y
fibrosis hepática
pueden establecer
el Dx. Los quistes
tienden a
mantener
conexiones con
nefronas
orifginarias.
Biopsia de hígado,
, pruebas de
diagnóstico
genético
- Descubrimiento
radiológico de
quistes. Uns
ecografía
abdominal o TC en
riñones
poliquísticos y
fibrosis hepática
pueden establecer
el Dx. Los quistes
tienden a
mantener
conexiones con
nefronas
orifginarias.
Biopsia de hígado,
, pruebas de
diagnóstico
genético
- Pronóstico
- Tasas de mortalidad más altas en
el primer año de vida, 50-80%
sobrevive hasta los 15 años
- Tasas de mortalidad más altas en
el primer año de vida, 50-80%
sobrevive hasta los 15 años
- Etiología
- EPQR
Autosómica
Dominante
- Etiología
- Se hereda AD con penetrancia
completa, mutación de dos
genes: PKD1 (en el cromosoma
16p13.3; 85% de los casos) y PKD2
(en el cromosoma 4q21-23; 15%
de los casos)
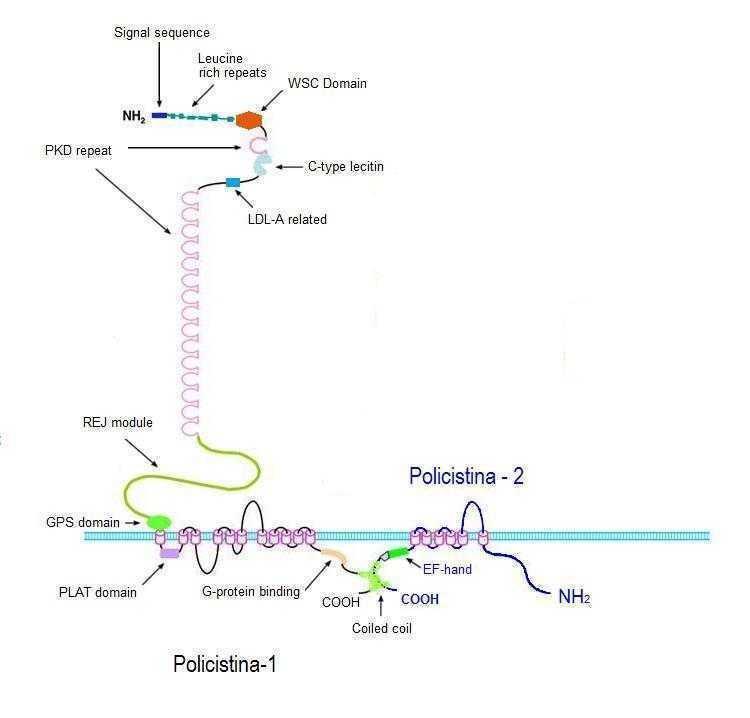
- PKD1 codifica-->
Poliquistina-1 (PC1),
PKD2 codifica --> poliquistina-2 (PC2)
- PKD1 codifica-->
Poliquistina-1 (PC1),
PKD2 codifica --> poliquistina-2 (PC2)
- Se hereda AD con penetrancia
completa, mutación de dos
genes: PKD1 (en el cromosoma
16p13.3; 85% de los casos) y PKD2
(en el cromosoma 4q21-23; 15%
de los casos)
- Epidemiología
- Factores preventivos/riesgo
- Incidencia de 1 : 400-1000,
desarrollo dependiente de la edad
--> afecta principalmente a los
adultos (20a y 30a). Afecta a
ambos géneros y todas las razas
por igual.
- Factores preventivos/riesgo
- Fisiopatología
- Complicaciones
- PKD1 tienden a una clínica más
severa, la mayoría desarrollan IRT,
desarrollo de un número mayor de
quistes a una edad más temprana
- modelo two-hit de quistogénesis o
doble golpe
- PKD1 tienden a una clínica más
severa, la mayoría desarrollan IRT,
desarrollo de un número mayor de
quistes a una edad más temprana
- PC1 y PC2 son (TRPP) de canales receptores
transitorios de potencial, ubicadas en los cilios
primarios. Ambas forman un complejo con al
función de regulación del calcio intracelular.
- Mecanismo de acción:
poliquistinas perciben y
traducen la estimulación
mecánica --> entrada de calcio
--> desencadena una mayor
liberación de calcio por el
retículo endoplásmico --> señal
de inhibición de proliferación
- Ausencia de poliquistinas --> falta de señal de
inhibición de proliferación --> incapacidad de
mantener la polaridad -- celular proliferación
incontrolada y generación de quístes, expresión de un
fenotipo secretorio y la remodelación de la matriz
extracelular
- Vías
implicadas:
AMPc,
mTOR, Wnt
canónica
- Ausencia de poliquistinas --> falta de señal de
inhibición de proliferación --> incapacidad de
mantener la polaridad -- celular proliferación
incontrolada y generación de quístes, expresión de un
fenotipo secretorio y la remodelación de la matriz
extracelular
- Mecanismo de acción:
poliquistinas perciben y
traducen la estimulación
mecánica --> entrada de calcio
--> desencadena una mayor
liberación de calcio por el
retículo endoplásmico --> señal
de inhibición de proliferación
- Complicaciones
- Manifestaciónes clínicas
- Manifestaciones
renales
- Desarrollo y crecimiento
quístico, disminuye la GFR,
Aumento de tamaño del
abdomen debido a que los
riñones están dilatados
Sangre en la orina Cálculos
renales Insuficiencia renal
Infecciones en las vías
urinarias o en los riñones
- Desarrollo y crecimiento
quístico, disminuye la GFR,
Aumento de tamaño del
abdomen debido a que los
riñones están dilatados
Sangre en la orina Cálculos
renales Insuficiencia renal
Infecciones en las vías
urinarias o en los riñones
- Manifestaciones
extrarrenales
- Presión arterial alta
Dolor de espalda o
lateral, dolor de
cabeza Una
sensación de
pesadez en el
abdomen
- Quistes del higado,
Aneurismas
cerebrales,
Prolapso de la
válvula mitral
- Quistes del higado,
Aneurismas
cerebrales,
Prolapso de la
válvula mitral
- Presión arterial alta
Dolor de espalda o
lateral, dolor de
cabeza Una
sensación de
pesadez en el
abdomen
- Manifestaciones
renales
- Diagnóstico
- pruebas radiológicas,
historia familiar
ecografía renal
(inocuidad y a su bajo
costo) - criterios
ultrasonográficos de
Ravine modificados
(no válidos para (TM
y RM)
- pruebas radiológicas,
historia familiar
ecografía renal
(inocuidad y a su bajo
costo) - criterios
ultrasonográficos de
Ravine modificados
(no válidos para (TM
y RM)
- Pronóstico
- peor progresión en hombres
con HTA mal controlada
ymutaciones en PKD1. 5%
fallecen debido a ruptura de
aneurisma cerebral
- peor progresión en hombres
con HTA mal controlada
ymutaciones en PKD1. 5%
fallecen debido a ruptura de
aneurisma cerebral
- Etiología
- Tratamiento
- No existe una cura,
el tratamiento es
sintomático
- No existe una cura,
el tratamiento es
sintomático
- Esporádica
- Muy rara
- Muy rara
- poliquistosis
Media attachments
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Want to create your own Mind Maps for free with GoConqr? Learn more.