20101906
Description
Mind Map by Lizeth Madueño, updated more than 1 year ago
|
|
Created by Lizeth Madueño
over 4 years ago
|
|
36. MALFORMACIONES CONGÉNITAS DEL
APARATO GENITAL FEMENINO
- Los defectos anatómicos del aparato reproductor femenino son frecuentes y son
consecuencia de mutaciones genéticas, detención del desarrollo o agresiones
ambientales que repercuten sobre las fases críticas del desarrollo embrionario.
- ANOMALÍAS DEL
CLÍTORIS
- CLITOROMEGALIA
- Cliteromegalia es sugestiva
de exposición del feto a un
exceso de andrógenos.
- El índice del clítoris
mayor de 10 mm2.
- El índice del clítoris
mayor de 10 mm2.
- Cliteromegalia es sugestiva
de exposición del feto a un
exceso de andrógenos.
- CLÍTORIS BÍFIDO
(1:480000).
- CLITOROMEGALIA
- MÉTODOS DIAGNÓSTICOS
- Historia clínica y examen físico.
Histerosalpingografía.
- Historia clínica y examen físico.
Histerosalpingografía.
- ANOMALÍAS DE
OVARIO
- OVARIO
SUPERMUNERARIO
- Glándula ectópica sin conexión
con los ligamentos anchos
uteroovárico o infundibulopélvico.
- LOCALIZACIÓN
- Retroperitoneo.
- Pelvis.
- Mesenterio de colon.
- Retroperitoneo.
- LOCALIZACIÓN
- Glándula ectópica sin conexión
con los ligamentos anchos
uteroovárico o infundibulopélvico.
- OVARIO
ACCESORIO
- Exceso de tejido ovárico
unido a un ovario normal.
- Exceso de tejido ovárico
unido a un ovario normal.
- OVARIO
SUPERMUNERARIO
- ANOMALÍAS EN LAS
TROMPAS DE FALOPIO
- Orificios accesorios
Agenesia completa
vestigios quísticos
- Orificios accesorios
Agenesia completa
vestigios quísticos
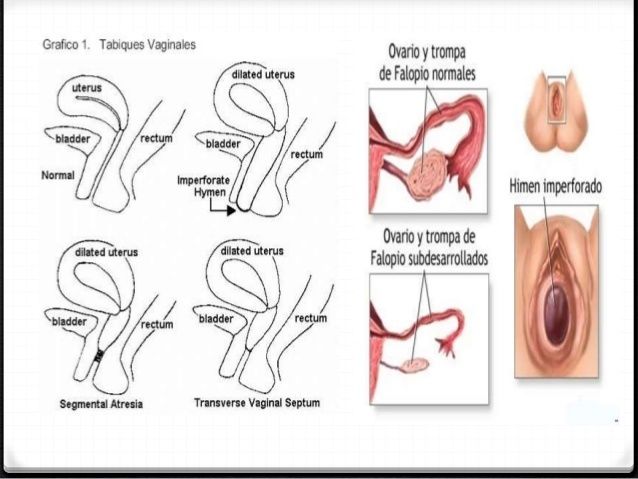
- TABIQUE VAGINAL
- LONGITUDINAL
- El tabique vaginal longitudinal es
secundario a la fusión lateral
deficiente y reabsorción incompleta
de los conductos de Muller.
- Parcial o abarcar toda la
longitud de la vagina.
- En la adolescencia hay menarquía
normal, con empeoramiento del dolor
vaginal y pélvico unilateral cada mes,
por obstrucción del conducto de salida.
- En la adolescencia hay menarquía
normal, con empeoramiento del dolor
vaginal y pélvico unilateral cada mes,
por obstrucción del conducto de salida.
- Parcial o abarcar toda la
longitud de la vagina.
- El tabique vaginal longitudinal es
secundario a la fusión lateral
deficiente y reabsorción incompleta
de los conductos de Muller.
- TRANSVERSO
- No se produce fusión de los conductos de Muller
ni de la canalización de la lámina vaginal.
- OBSTRUCTIVO
- Con acumulación de moco
o sangre menstrual.
- Con acumulación de moco
o sangre menstrual.
- NO OBSTRUCTIVO
- Salida de moco y sangre.
- Salida de moco y sangre.
- Tercio superior 46%
Tercio medio 36%
Tercio inferior 19%
- El espesor septal varía pero
por lo regular es delgado (1
cm), los más gruesos llegan a
medir hasta 5 o 6 cm y
tienden a ubicarse más cerca
del cuello uterino.
- El espesor septal varía pero
por lo regular es delgado (1
cm), los más gruesos llegan a
medir hasta 5 o 6 cm y
tienden a ubicarse más cerca
del cuello uterino.
- OBSTRUCTIVO
- No se produce fusión de los conductos de Muller
ni de la canalización de la lámina vaginal.
- LONGITUDINAL
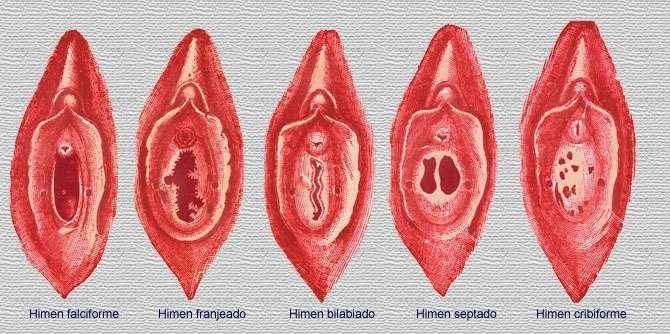
- DEFECTOS DEL HIMEN
- Microperforada, tabicada,
cribiforme e imperforada.
- Los pacientes con himen microperforado,
cribiforme o tabicado presentan irregularidades
menstruales, dificultad para introducir un
tampón o problemas durante el coito.
- Los pacientes con himen microperforado,
cribiforme o tabicado presentan irregularidades
menstruales, dificultad para introducir un
tampón o problemas durante el coito.
- Microperforada, tabicada,
cribiforme e imperforada.
- AMBIGUEDAD CONGÉNITA
DEL APARATO GENITAL
- Anomalías de la maduración genética gonadal.
- DISGENESIA GONADAL
- Resultados de la no disyunción de los cromosomas
de los padres; provocando un desarrollo gonadal
anormal, con presencia de estrías gonadales.
- 40-50% de los pacientes con disgenesia gonadal
el cariotipo es 45X y este trastorno se conoce
como síndrome de Turner.
- Síndrome se reconoce desde la infancia,
pero algunas pacientes se diagnostican
hasta la adolescencia, donde manifiestan
talla baja, amenorrea primaria,
hipogonadismo hipergonadotrópico y
genitales femeninos prepuberales.
- Síndrome se reconoce desde la infancia,
pero algunas pacientes se diagnostican
hasta la adolescencia, donde manifiestan
talla baja, amenorrea primaria,
hipogonadismo hipergonadotrópico y
genitales femeninos prepuberales.
- 40-50% de los pacientes con disgenesia gonadal
el cariotipo es 45X y este trastorno se conoce
como síndrome de Turner.
- Resultados de la no disyunción de los cromosomas
de los padres; provocando un desarrollo gonadal
anormal, con presencia de estrías gonadales.
- HERMAFRODITISMO VERDADERO
- Hermafrodita verdadero posee
tejido tanto ovárico como testicular.
- El caritipo más frecuente es 46
XX, seguido de 46 XX/46 XY.
- Fenotipo de una hermafrodita verdadero 46
XX comprende un ovotestículo unilateral
con un ovario o testículo contralateral o
bien ovotestículos bilaterales.
- Por lo general los genitales externos son
ambiguos y poco masculinizados por la
cantidad insuficiente de testosterona.
- Por lo general los genitales externos son
ambiguos y poco masculinizados por la
cantidad insuficiente de testosterona.
- Fenotipo de una hermafrodita verdadero 46
XX comprende un ovotestículo unilateral
con un ovario o testículo contralateral o
bien ovotestículos bilaterales.
- El caritipo más frecuente es 46
XX, seguido de 46 XX/46 XY.
- Hermafrodita verdadero posee
tejido tanto ovárico como testicular.
- PSEUDOHERMAFRODITISMO
MASCULINO
- La exposición insuficiente a los andrógenos en
el feto destinado a ser varón, provoca
pseudohermafroditismo masculino. El
cariotipo es 46 XY y existen testículos.
- Estos pacientes casi siempre son estériles
a causa de una espermatogénesis
anormal y poseen un falo pequeño o
insuficiente para la función sexual.
- TRATAMIENTO
- Administración de estrógenos hasta lograr
una concentración fisiológica, ya sea por
dilatación o vaginoplastía quirúrgica.
- Administración de estrógenos hasta lograr
una concentración fisiológica, ya sea por
dilatación o vaginoplastía quirúrgica.
- TRATAMIENTO
- Estos pacientes casi siempre son estériles
a causa de una espermatogénesis
anormal y poseen un falo pequeño o
insuficiente para la función sexual.
- La exposición insuficiente a los andrógenos en
el feto destinado a ser varón, provoca
pseudohermafroditismo masculino. El
cariotipo es 46 XY y existen testículos.
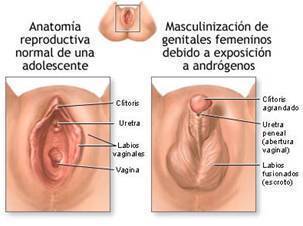
- PSEUDOHERMATRODITISMO FEMENINO
- La discordancia entre el sexo gonadal (46 XX) y
el aspecto de los genitales externos
(masculinizados) es secundario a la exposición
excesiva del embrión o feto a los andrógenos.
- Existen ovarios y estructuras ductales
internas femeninas como útero, cuello
uterino y tercio superior de la vagina,
siento potencialmente fértiles.
- ASPECTO
- Cliteromegalia moderada hasta la
formación de un falo con uretra
peniana. Pliegues labioescrotales
y seno urogenital.
- TRATAMIENTO
- La corrección quirúrgica debe abarcar a
las anomalías de estas estructuras para
obtener un buen resultado estético y
una función sexual adecuada.
- La corrección quirúrgica debe abarcar a
las anomalías de estas estructuras para
obtener un buen resultado estético y
una función sexual adecuada.
- TRATAMIENTO
- Cliteromegalia moderada hasta la
formación de un falo con uretra
peniana. Pliegues labioescrotales
y seno urogenital.
- ASPECTO
- Existen ovarios y estructuras ductales
internas femeninas como útero, cuello
uterino y tercio superior de la vagina,
siento potencialmente fértiles.
- La discordancia entre el sexo gonadal (46 XX) y
el aspecto de los genitales externos
(masculinizados) es secundario a la exposición
excesiva del embrión o feto a los andrógenos.
- DISGENESIA GONADAL
- Anomalías de la maduración genética gonadal.
- ANOMALÍAS DE LOS CONDUCTOS DE MULLER
- Las anomalías del útero pueden ser congénitas o
adquiridas y se acompañan de anomalías del ciclo
menstrual, dolor pélvico, infertilidad o aborto.
- Los defectos mullerianos se acompañan
de anomalías renales en un 30-50%:
- Agenesia renal unilateral
Hipoplasia renal intensa
Riñón en herradura.
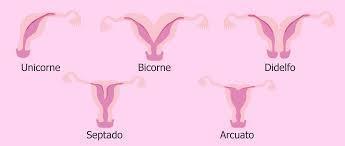
- CLASE I: Hipoplasia o
agenesia segmentaria de los
conductos de Muller.
- CLASE II: Útero unicorne.
- Clase III: Útero didelfo.
- Clase IV: Útero bicorne.
- Clase V: Útero tabicado.
- Clase VI: Útero arqueado.
- CLASE I: Hipoplasia o
agenesia segmentaria de los
conductos de Muller.
- Agenesia renal unilateral
Hipoplasia renal intensa
Riñón en herradura.
- Los defectos mullerianos se acompañan
de anomalías renales en un 30-50%:
- Las anomalías del útero pueden ser congénitas o
adquiridas y se acompañan de anomalías del ciclo
menstrual, dolor pélvico, infertilidad o aborto.
- ANOMALÍAS DEL
CLÍTORIS
Media attachments
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Want to create your own Mind Maps for free with GoConqr? Learn more.