3278772
Description
Mind Map by Luciani Palacios Garcia, updated more than 1 year ago
|
|
Created by Luciani Palacios Garcia
over 9 years ago
|
|
FARMACOGENÉTICA: HIPERTERMIA MALIGNA
- "Hiperexia maligna" / "Fiebre maligna" / "Fiebre anestésica"
- Enfermedad genética autosómica dominante que afecta
al músculo esquelético.
- Enfermedad genética autosómica dominante que afecta
al músculo esquelético.
- MECANISMO DE ACCIÓN
- Este proceso es el resultado de una anormalidad debido al incremento en la salida del calcio del
retículo sarcoplasmico, el cual es frecuentemente causado por una mutación heredada en el gen
receptor de la rianodina ( RYR1), que se encuentra en la membrana del retículo sarcoplasmico.
- La prueba ideal para la determinación de la susceptibilidad a la hipertermia maligna es la prueba de
contractura a Cafeína-Halotano.
- Las mutaciones del gen de la rianodina se encuentran por lo menos en el 25 % de los individuos
susceptibles a hipertermia maligna en los estados unidos.
- La HM obedece a una alteración en la regulación del calcio en la célula muscular esquelética principalmente y
luego en la cardiaca. Cuando se despolariza la membrana sale calcio del retículo sarcoplásmico a través de los
canales del calcio y los receptores tipo 1 de la rianodina (RYR1). Este canal no es un canal sencillo y hay proteínas
que regulan la apertura y cierre del canal. Además existe otro canal que es el canal receptor de la
dehidropiridina (DHPR).
- El 70% de las mutaciones están en el RYR1, hay otros genes como el de la DHPR que ocupan el otro
30%.
- La secuencia del RYR1, se aprecia que es un gen muy grande, tiene más de 150.000 pares de bases,
se estudiaron los 3 puntos calientes en los exones 2-17, 39-46 y 90- 104, los exones son puntos del
gen que codifican las proteínas, se apreciaron algunas mutaciones y otros polimorfismos.
- El gen RYR1 en el humano es autosomico dominante y esta ligada en un poco mas del 50% de las
familias suceptibles a contraer la enfermedad.
- Este proceso es el resultado de una anormalidad debido al incremento en la salida del calcio del
retículo sarcoplasmico, el cual es frecuentemente causado por una mutación heredada en el gen
receptor de la rianodina ( RYR1), que se encuentra en la membrana del retículo sarcoplasmico.
- SINTOMATOLOGÍA
- Aumento rápido de la temperatura a 105 grados F (40.5 º C) o mayor
- Tensión y rigidez muscular.
- Orina de color pardo oscuro.
- Dolor muscular sin que se presente un ejercicio obvio que explique los músculos adoloridos.
- Sangrado
- Aumento rápido de la temperatura a 105 grados F (40.5 º C) o mayor
- TRATAMIENTO
- Serie de medidas encaminadas a
descontinuar el proceder anestésico,
combatir la hipoxemia y la acidosis,
controlar la T° corporal y contrarrestar la
posibles complicaciones.
- Interrupir indemidatamente la anestesia y la cirugía.
- Hiperventilar al paciente con O2 al 100 %.
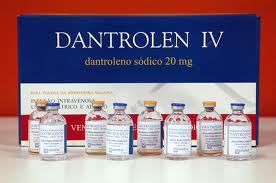
- Administrar dantroleno de 1 a 2,5 mg/kg intravenosa (IV), dosis que deberá repetirse cada 5 ó 10 min
hasta una dosis total de 10 mg/kg, aunque debe administrarse más si persisten los síntomas.
- Administrar Na CO2 de 2 a 4 mEq/kg, IV, y en dosis mayores si así lo sugiere el pH arterial y la Pa
CO2 .
- Controlar la temperatura corporal mediante varias formas de enfriamiento activo.
- Mantener la diuresis con manitol 25 g, IV, furosemida 20 mg, IV y un aporte de líquido IV abundante.
- Mantener un cuidadoso monitoreo del paciente y continuar el tratamiento hasta que él esté estable
y posteriormente hasta que desaparezca el riesgo de nuevos episodios.
- El dantroleno es un relajante muscular de acción rápida que impide la liberación
de iones de Ca desde el retículo sarcoplásmico, de esta manera disminuye la
espasticidad, se normaliza la función muscular y, finalmente, se revierten los
cambios metabólicos. Anteriormente la mortalidad por HM era casi del 80 %,
pero con más información y la disponibilidad del dantroleno, la mortalidad ha
bajado al 10 % en los países desarrollados.
- Serie de medidas encaminadas a
descontinuar el proceder anestésico,
combatir la hipoxemia y la acidosis,
controlar la T° corporal y contrarrestar la
posibles complicaciones.
- ETIOLOGÍA
- GENÉTICO
- Transtorno autosómico dominantes
- Mutación en:
- GEN RYRI (codifica canal interc. de Ca)
- GEN CACNLIA3 ( codifica sub unidad α 1 de canal de Ca)
- GEN RYRI (codifica canal interc. de Ca)
- Mutación en:
- Transtorno autosómico dominantes
- FACTORES CUALIFICANTES
- Estrés físico o emocional, ansiedad excesiva, hiperactividad
adrenérgica, temperatura ambiental elevada, hipertermia
preoperatoria
- Estrés físico o emocional, ansiedad excesiva, hiperactividad
adrenérgica, temperatura ambiental elevada, hipertermia
preoperatoria
- FÁRMACOS DESENCADENANTES
- En respuesta a un producto miorrelajante (SUCCINILCOLINA)
- Anestésico inhalado (HALONATO)
- Fármacos conocidos desencadenantes de la HM
- En respuesta a un producto miorrelajante (SUCCINILCOLINA)
- GENÉTICO
Media attachments
{kind=link}
{kind=link}
{kind=link}
Want to create your own Mind Maps for free with GoConqr? Learn more.