6715785
Nefropatía Poliquística de la
infantil.
- Concepto
- Es una enfermedad genética ,forma
frecuentemente grave de enfermedad renal
poliquística, que consiste en la aparición
progresiva de lesiones quísticas en los
riñones, que remplazan el parénquima
renal, lo que conduce a enfermedad renal
crónica terminal.
- Es una enfermedad genética ,forma
frecuentemente grave de enfermedad renal
poliquística, que consiste en la aparición
progresiva de lesiones quísticas en los
riñones, que remplazan el parénquima
renal, lo que conduce a enfermedad renal
crónica terminal.
- Impacto
- Incidencia
- de 1:20.000
nacidos vivos
- de 1:20.000
nacidos vivos
- Prevalencia
- 1:400 hasta 1:1.000
- 1:400 hasta 1:1.000
- Incidencia
- Etiología
- Se produce por
mutaciones en
varios loci
humanos.
- Se produce por
mutaciones en
varios loci
humanos.
- Fisiopatología
- Hay una dilatación variable de los túbulos colectores y de los ductos biliares. Es por ello típica la
aparición de unos riñones voluminosos con numerosos microquistes inferiores a tres milímetros,
que corresponden a túbulos colectores dilatados por fluido acumulado en su interior. Los
numerosos quistes ejercen un efecto compresivo sobre el parénquima sano, dando lugar a la
progresiva destrucción de nefronas (lo que conlleva a falla renal). Esto justifica la escasa diuresis
intrauterina del feto, que conlleva un oligohidramnios y consecuentemente una hipoplasia
pulmonar.
- Hay una dilatación variable de los túbulos colectores y de los ductos biliares. Es por ello típica la
aparición de unos riñones voluminosos con numerosos microquistes inferiores a tres milímetros,
que corresponden a túbulos colectores dilatados por fluido acumulado en su interior. Los
numerosos quistes ejercen un efecto compresivo sobre el parénquima sano, dando lugar a la
progresiva destrucción de nefronas (lo que conlleva a falla renal). Esto justifica la escasa diuresis
intrauterina del feto, que conlleva un oligohidramnios y consecuentemente una hipoplasia
pulmonar.
- Manifestaciones clínicas
- Los casos más graves pueden
manifestarse por oligohidramnios
antes del nacimiento
- hipoplasia pulmonar
- hipoplasia pulmonar
- masas renales palpables, dificultad respiratoria y, por
el gran tamaño de los riñones con compresión
diafragmática y del estómago, pueden mostrar
dificultad para la alimentación.
- hipertensión arterial, que se presentará entre 55 % y 75 % de los casos
- Complicaciones
- Fibrosis hepática
- Insuficiencia renal
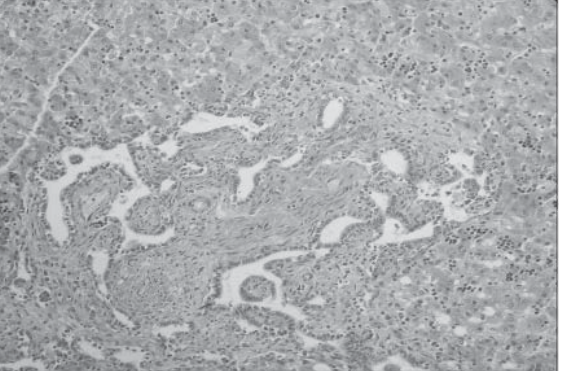
- Fibrosis de los espacios porta con dilatación y
distorsión de los conductos biliares E 400X
- Insuficiencia renal
- dilatación quística de los conductos biliares
- Síndrome de Caroli
- Riesgo a colangitis recurrente
- Riesgo a colangitis recurrente
- Síndrome de Caroli
- Debido al bajo gasto urinario fetal,
puede desarrollar la secuencia de Potter
(hipoplasia pulmonar, fascies típicas y
anomalías en extremidades)
- Fibrosis hepática
- hipertensión portal
- Los casos más graves pueden
manifestarse por oligohidramnios
antes del nacimiento
- Genética
- -Mutaciones gen PKHD1 ( se espresa en el
riñon adulto y fetal, hígado y páncreas) en
cromosoma 6p21-p23
- -Gen que codifica la FIBROCISTINA que se localiza
en el cilio primario de las células tubulares.
- Podría participar en la diferenciación del túbulo colector y biliar
- Podría participar en la diferenciación del túbulo colector y biliar
- -Gen que codifica la FIBROCISTINA que se localiza
en el cilio primario de las células tubulares.
- Autosómica recesiva
- Heterocigotos compuestos
- -Mutaciones gen PKHD1 ( se espresa en el
riñon adulto y fetal, hígado y páncreas) en
cromosoma 6p21-p23
- Demografía
- Género
- No se ha encontrado relación con el género
- No se ha encontrado relación con el género
- Edad
- La forma perinatal y neonatal son las más frecuentes. Perinatal hasta los 2 años
- La forma perinatal y neonatal son las más frecuentes. Perinatal hasta los 2 años
- Género
- Morfología Microscópica
- Dilatación cilíndrica o , con
menor frecuencia sacular, de
todos los conductos
colectores. riño (microquistes
menores a 4 mm de
diámetro) radiados desde la
médula hacia la corteza
- Quistes revestidos con células cúbicas
que reflejan su origen en los túbulos
colectores
- quistes que están agrandando las estructuras tubulares, que llegan de la superficie cortical y se extienden
dentro de la medula
- quistes que están agrandando las estructuras tubulares, que llegan de la superficie cortical y se extienden
dentro de la medula
- Hay pequeñas áreas de
parénquima normal en la
corteza sin evidencia de
displasia, aunque se han
reportado células
musculares lisas
alrededor de los quistes.
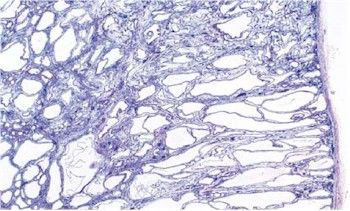
- Riñón en la Enfermedad
Poliquística Autosómica
recesiv. Obsérvese los
quistes de disposición
longitudinal, constituidos por
túbulos colectores. Se
identifican estructuras
glomerulares conservadas
100X.
- Dilatación quística de los túbulos
colectores en la enfermedad
poliquística autosómica recesiva. 450X
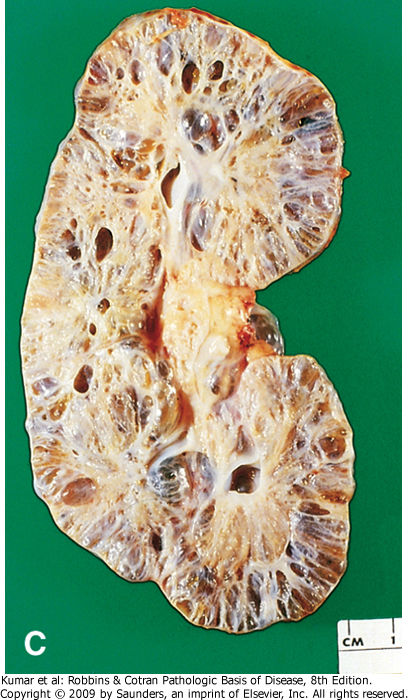
- Se observa una profusión de quistes que sustituyen
completamente el parénquima renal con algunos glomérulos
incluidos (Hematoxilina-Eosina, x20).
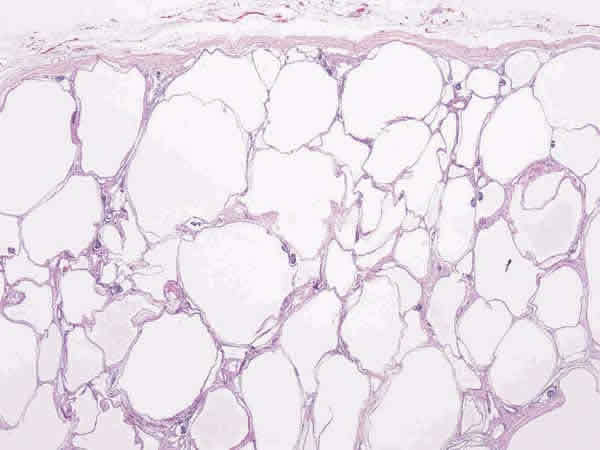
- Corte de riñón en el que see visualizan muchos
quistes pequeños en la corteza y médula renal.
Canales alargados y dilatados reemplazando
corteza, se alcanzan a percibir quistes que están
revestidos por celular cubicas
- Dilatación cilíndrica o , con
menor frecuencia sacular, de
todos los conductos
colectores. riño (microquistes
menores a 4 mm de
diámetro) radiados desde la
médula hacia la corteza
- Morfología Macroscópica
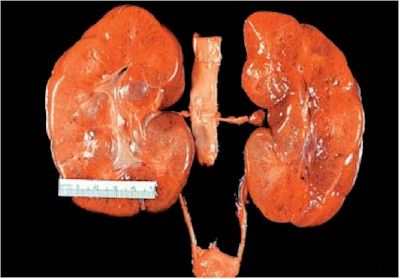
- Riñones quísticos aumentados de tamaño en el nacimiento
- Aspecto externo normal
- Al corte, muchos quistes pequeños en la corteza y la médula dan aspecto de ESPONJA
- Canales alargados y dilatados en ángulo recto con
la superficie cortical, reemplazan completamente
la médula y la corteza.
- compromiso renal simétrico y bilateral
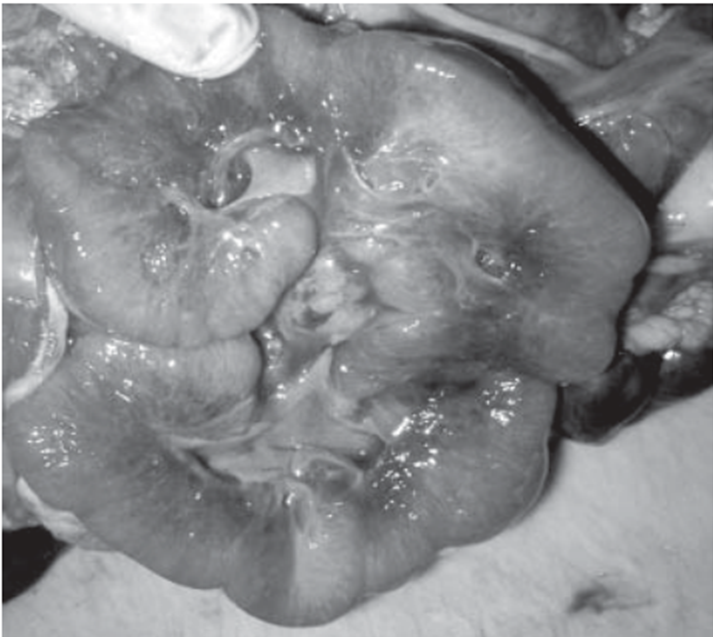
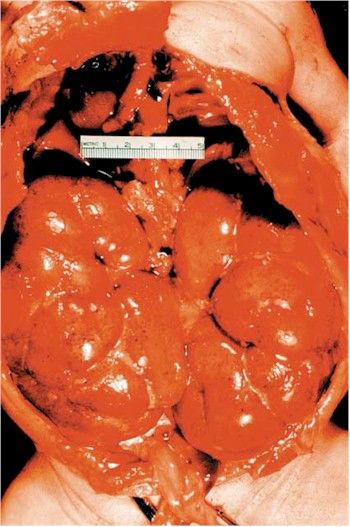
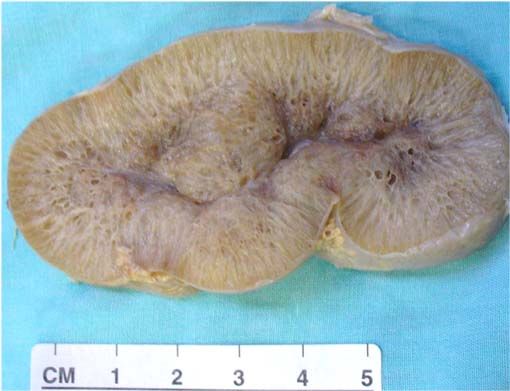
- Riñón en la enfermedad poliquística autosómica recesiva. El riñón se encuentra muy aumentado de
tamaño y al corte se observan numerosos quistes delgados y elongados.
- Los riñones están mostrando in situ
que ocupan casi la cavidad
abdominal completa de un niño
recién nacido
- Riñones quísticos aumentados de tamaño en el nacimiento
- Pronóstico
- Las formas Lactante y juvenil son las que sobreviven.--> aunque desarrollan fibrosis hepática
Mientras que la perinatal y neonatal apesar de que son
las MÁS frecuentes, tienen peor pronóstico
- Más de la mitad de los pacientes con enfermedad renal poliquística autosómica recesiva/fibrosis
hepática congénita (EPAR/FHC) requieren trasplante renal antes de alcanzar los 20 años de edad
- 30% de los neonatos afectados fallece a causa de insuficiencia respiratoria
- casi todos los pacientes con mutaciones bialélicas truncadas o de marco de lectura abierto tienen un fenotipo
más grave de la enfermedad
- sobrevida de los
pacientes, la tasa
anual es del 71-75%
a los 10 años y del
66% a los 15 años
- Las formas Lactante y juvenil son las que sobreviven.--> aunque desarrollan fibrosis hepática
Mientras que la perinatal y neonatal apesar de que son
las MÁS frecuentes, tienen peor pronóstico
- Tratamiento
- estabilización de la función
respiratoria, mediante
ventilación mecánica
- evaluación de la función
respiratoria, pulsoximetría,
radiografía de tórax y
exámenes paraclínicos
pertinentes según el caso
- Si se presenta oliguria o
anuria, se debe iniciar
diálisis peritoneal
- nefrectomía unilateral o
bilateral según el
compromiso
- hipertensión con
inhibidores de la IECA o
ARA II
- El tratamiento definitivo es
el transplante renal, pero
debe valorarse el grado de
daño hepático existent
- estabilización de la función
respiratoria, mediante
ventilación mecánica
- Diagnóstico
- sospecharse prenatalmente por
oligohidramnios y vejiga vacía.
- Ultrasonido materno-fetal
- agrandamiento renal masivo, hiperecogenicidad del
parénquima, no diferenciación córtico-medular,
pequeños quistes menores de 2 cm de diámetro y
aumento de la ecogenicidad hepática.
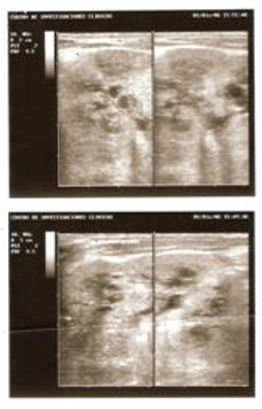
- Ultrasonido renal en un paciente de 26 días de nacido que
muestra riñones muy aumentados de tamaño (9 cm el derecho y
8 cm el izquierdo) y pequeños quistes con diámetro entre 1,5 y
4,0 mm
- Ultrasonido renal en un paciente de 26 días de nacido que
muestra riñones muy aumentados de tamaño (9 cm el derecho y
8 cm el izquierdo) y pequeños quistes con diámetro entre 1,5 y
4,0 mm
- agrandamiento renal masivo, hiperecogenicidad del
parénquima, no diferenciación córtico-medular,
pequeños quistes menores de 2 cm de diámetro y
aumento de la ecogenicidad hepática.
- diagnóstico prenatal
es posible utilizando
análisis del ADN
- de células fetales, por
amniocentesis (entre la semana 15 y
18 de gestación) o a la semana 12 de
gestación, por biopsia de
vellosidad coriónica
- de células fetales, por
amniocentesis (entre la semana 15 y
18 de gestación) o a la semana 12 de
gestación, por biopsia de
vellosidad coriónica
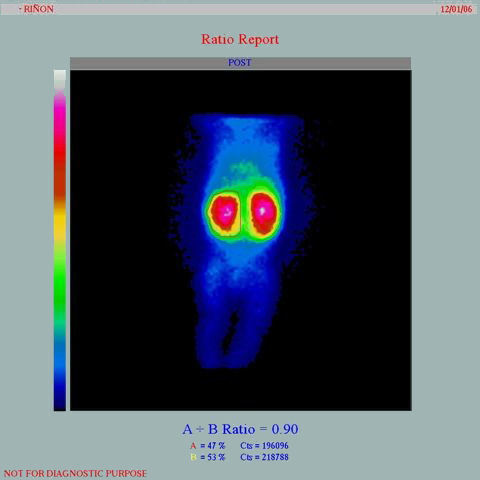
- estudio gammagráfico con ácido dimercaptosuccínico
marcado con tecnecio 99 (Tc99m-DMSA) muestra captación
en parches del radioisótopo, especialmente en los polos
renales
- Estudio gammagráfico estático (DMSA), que muestra el gran tamaño de los riñones
con hipocaptación bilateral de ambos polos, que se hace más evidente en el polo
superior izquierdo en esta figura.
- Estudio gammagráfico estático (DMSA), que muestra el gran tamaño de los riñones
con hipocaptación bilateral de ambos polos, que se hace más evidente en el polo
superior izquierdo en esta figura.
- El diagnóstico puede ser realizado en el periodo intrauterino, neonatal o en los primeros meses de
vida, por medio de una ecografía renal que evidencie un aumento difuso del volumen renal bilateral.
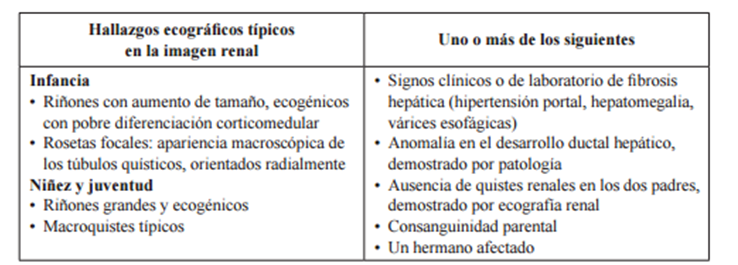
- Criterios diagnósticos:
- Criterios diagnósticos:
- Aunque la fibrosis hepática está histológicamente presente desde el nacimiento, los hallazgos
clínicos, radiológicos o paraclínicos pueden estar ausentes en el momento del diagnóstico
- encontrándose hallazgos ecográficos
en las semanas 14 a 17 de gestación
en los casos más severos, pero de
manera general, entre las 24 y 30
semanas, siendo tardíos para poder
realizar un diagnóstico prenata
- encontrándose hallazgos ecográficos
en las semanas 14 a 17 de gestación
en los casos más severos, pero de
manera general, entre las 24 y 30
semanas, siendo tardíos para poder
realizar un diagnóstico prenata
- transaminasas,
bilirrubinas séricas,
albumina sérica,
tiempos de
coagulación y
hemograma.
- Métodos
especiales
- sospecharse prenatalmente por
oligohidramnios y vejiga vacía.
- Métodos
especiales
- Clasificación
- Perinatal
- Presentación al nacimiento
- Riñones muy voluminosos;
hipoplasia pulmonar con
afectación hepática mínima;
quistes en 90 % de los túbulos y
muerte neonatal.
- Riñones muy voluminosos;
hipoplasia pulmonar con
afectación hepática mínima;
quistes en 90 % de los túbulos y
muerte neonatal.
- Presentación al nacimiento
- Neonatal
- Presentación durante el primer mes de vida
- Gran nefromegalia; leve afectación hepática;
insuficiencia renal progresiva y precoz, y
dilataciones quísticas en 65 % de los túbulos.
- Gran nefromegalia; leve afectación hepática;
insuficiencia renal progresiva y precoz, y
dilataciones quísticas en 65 % de los túbulos.
- Presentación durante el primer mes de vida
- Lactante
- Forma lactante y juvenil sobreviven a la
infancia, desarrollan fibrosis periportal
blanda y proliferación de conductillos
biliares
- 3 y 6 meses de edad
- Fibrosis hepática congénita( incluso se ha descrito en ausencia de riñones
poliquísticos y a veces en Nefropatía Poliquística del adulto) Dilataciones quísticas en
25% de los túbulos
- Hipertensión portal con esplenomegalia
- Hipertensión portal con esplenomegalia
- Fibrosis hepática congénita( incluso se ha descrito en ausencia de riñones
poliquísticos y a veces en Nefropatía Poliquística del adulto) Dilataciones quísticas en
25% de los túbulos
- 3 y 6 meses de edad
- Forma lactante y juvenil sobreviven a la
infancia, desarrollan fibrosis periportal
blanda y proliferación de conductillos
biliares
- Juvenil
- Después del año de edad
- hepatoesplenomegalia; grandes alteraciones hepáticas
e insuficiencia renal con quistes en 10 % de los túbulos
- hepatoesplenomegalia; grandes alteraciones hepáticas
e insuficiencia renal con quistes en 10 % de los túbulos
- Después del año de edad
- Perinatal
- Generalidades
- -Manifestaciones graves en el nacimiento
-Hígado con quistes asociados a fibrosis
portal y proliferación de las vías biliares
portales
- La enfermedad renal poliquística
autosómica recesiva (ERPAR) es una entidad
rara en la población neonatal
- Es un o de los dos subtipos de nefropatía políquística: que son la
autosómica dominante ( de adultos), y la autosómica recesiva
(Infantil))
- -Manifestaciones graves en el nacimiento
-Hígado con quistes asociados a fibrosis
portal y proliferación de las vías biliares
portales
Media attachments
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Want to create your own Mind Maps for free with GoConqr? Learn more.