1090121
Description
Mind Map by Kristi Brogden, updated more than 1 year ago
|
|
Created by Kristi Brogden
over 11 years ago
|
|
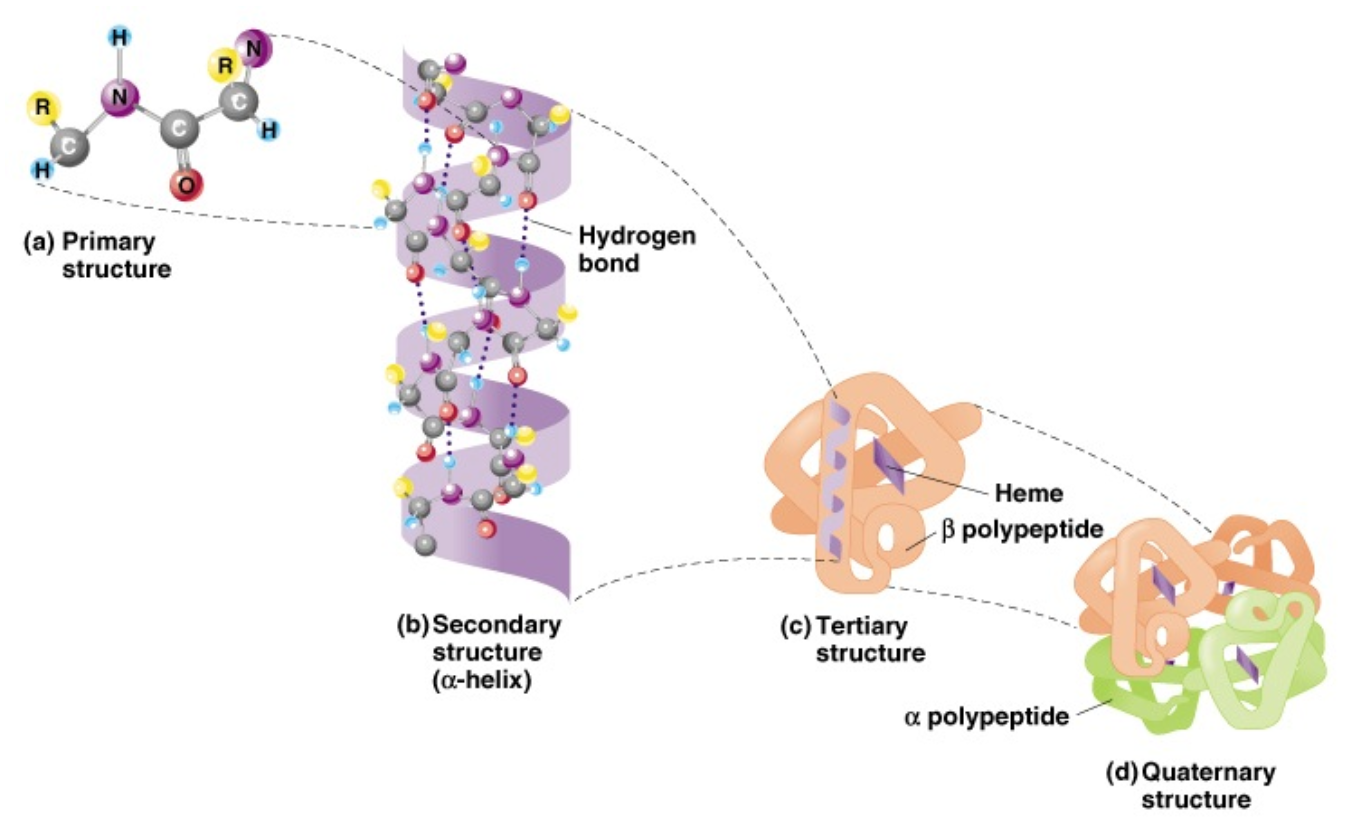
Protein structure (and function)
- Primary structure
- How do we find primary structure?
- mostly inferred from DNA sequence
- Prediction is based on the
need for amino acids to be in a
certain position in order to
achieve the desired structure
- Protein databases can provide
a wealth of predictions
- Sequence alignment
- Domain composition
- post-translational modifications
- protein-protein interactions
- structure
- etc.
- Sequence alignment
- Protein databases can provide
a wealth of predictions
- Prediction is based on the
need for amino acids to be in a
certain position in order to
achieve the desired structure
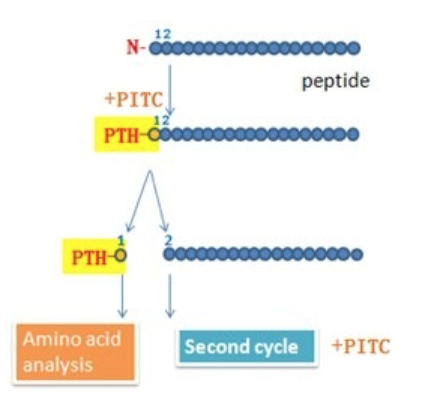
- Can be obtained directly by amino acid
sequencing using edman degradation
- Or using mass spectrometry
- mostly inferred from DNA sequence
- How do we find primary structure?
- Secondary structure
- Alpha-helix
- Space-filling
- Backbone
- Sticks
- Ribbon
- Space-filling
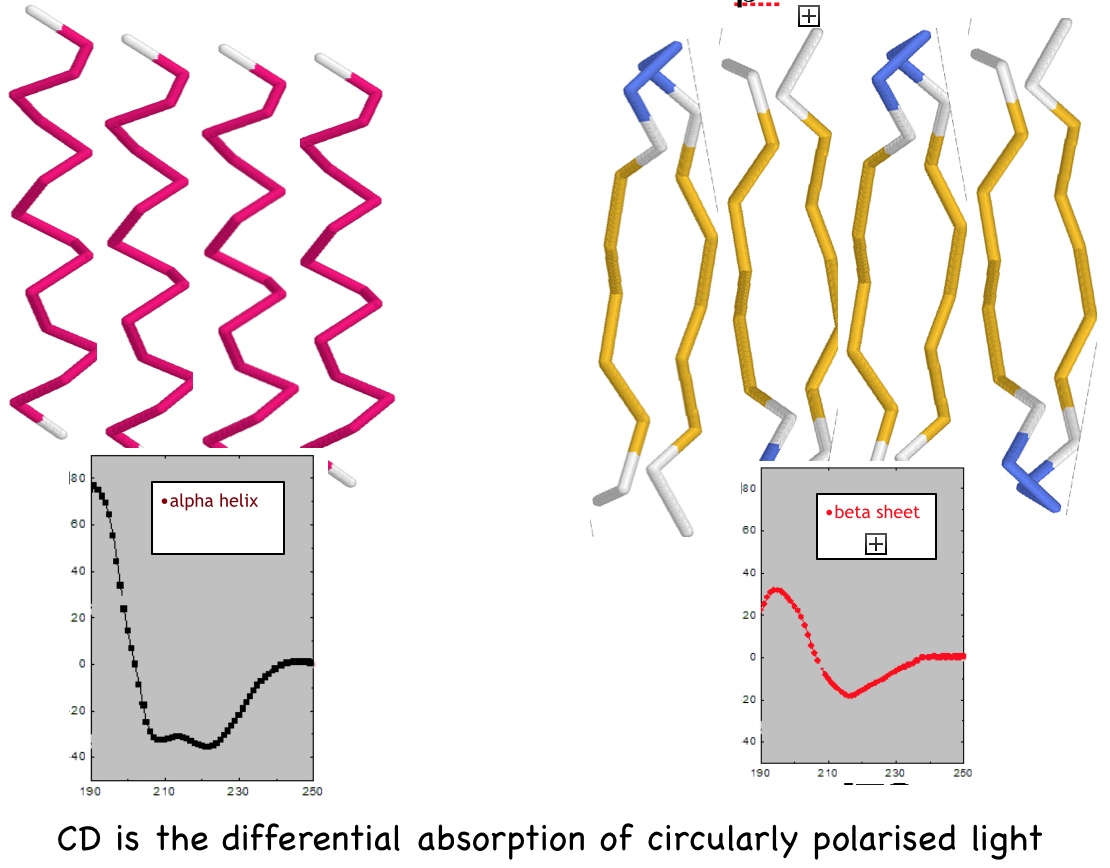
- Experimental determination of
Protein Secondary Structure by
Circular Dichroism (CD)
- CD spectroscopy in the "far-uv"
spectral region (190-250 nm)
reveals secondary structure.
- Alpha-helix, beta-sheet, and
random coil each give a
characteristic shape of CD
spectrum.
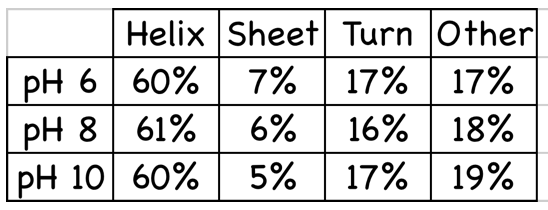
- The fraction of each secondary
structure type in any protein can
be calculated from its far-uv CD
spectrum.
- This gives a % for each
secondary structure element but
no information on arrangement.
- CD spectroscopy in the "far-uv"
spectral region (190-250 nm)
reveals secondary structure.
- Alpha-helix
- Tertiary structure
- The way in which individual secondary
structural elements; α-helices, β-sheets
and random coil, pack together within a
protein and between sub-domains of a
protein
- Information on tertiary Structure from CD
- The CD spectrum of a protein in the
"near-uv" spectral region (250-350 nm) gives
some information on tertiary structure
- CD signals of aromatic amino acids and
disulfide bonds, are sensitive to the overall
tertiary structure of the protein.
- Their absorbance is affected by the local
‘environment’ and can be observed
dynamically.
- Protein unfolding or
‘melting’ can be followed
by CD at different
temperatures.
- The CD spectrum of a protein in the
"near-uv" spectral region (250-350 nm) gives
some information on tertiary structure
- The way in which individual secondary
structural elements; α-helices, β-sheets
and random coil, pack together within a
protein and between sub-domains of a
protein
- Quaternary structure
- Quaternary structure is the
relationship between individual
proteins in a multimeric complex
- Quaternary structure is the
relationship between individual
proteins in a multimeric complex
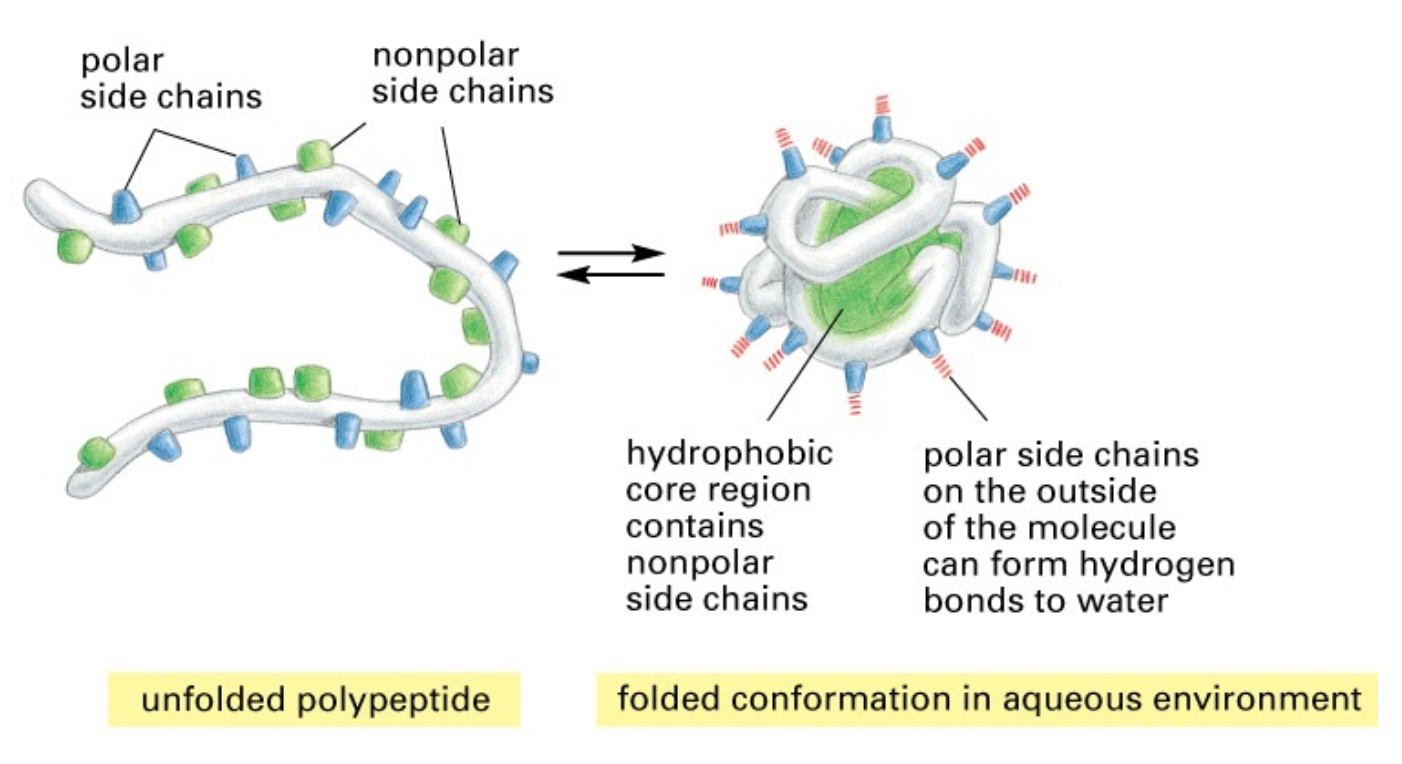
- Folding of a peptide into a protein
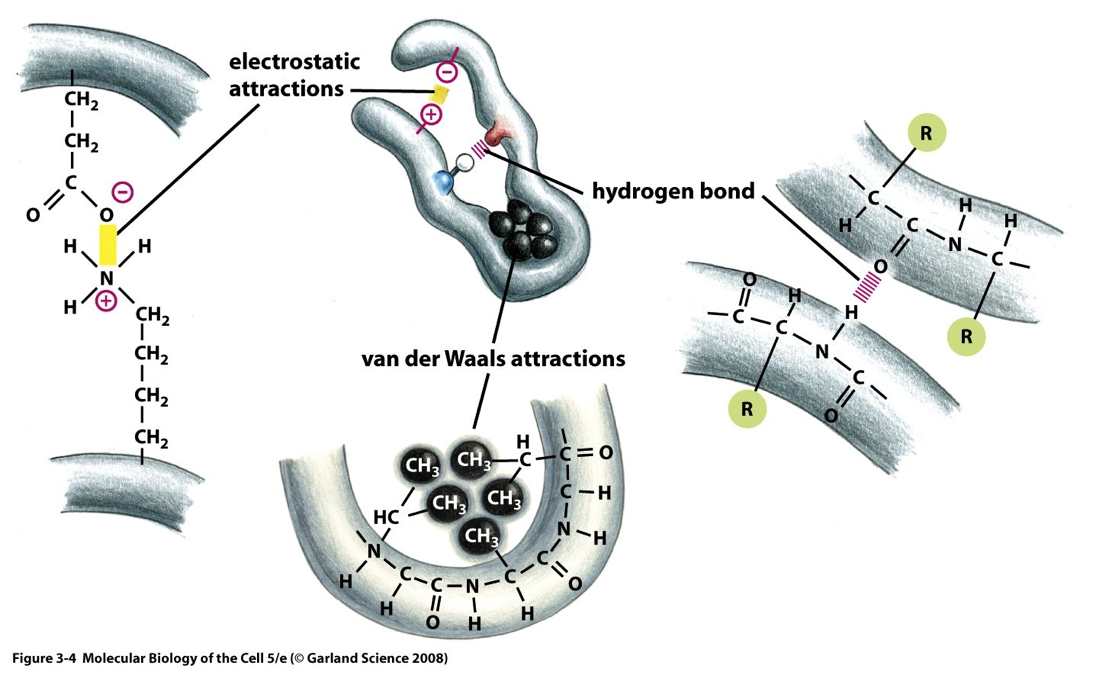
- Proteins are held together by different ionic interactions
- Ionic interactions
- Attraction between +ve and -ve charged ions
- Attraction between +ve and -ve charged ions
- Van der waals
- Short range weak electrical attraction and repulsion
- Short range weak electrical attraction and repulsion
- Hydrogen bonds
- Involve a H shared between O and N atoms
- Involve a H shared between O and N atoms
- Ionic interactions
- Other techniques
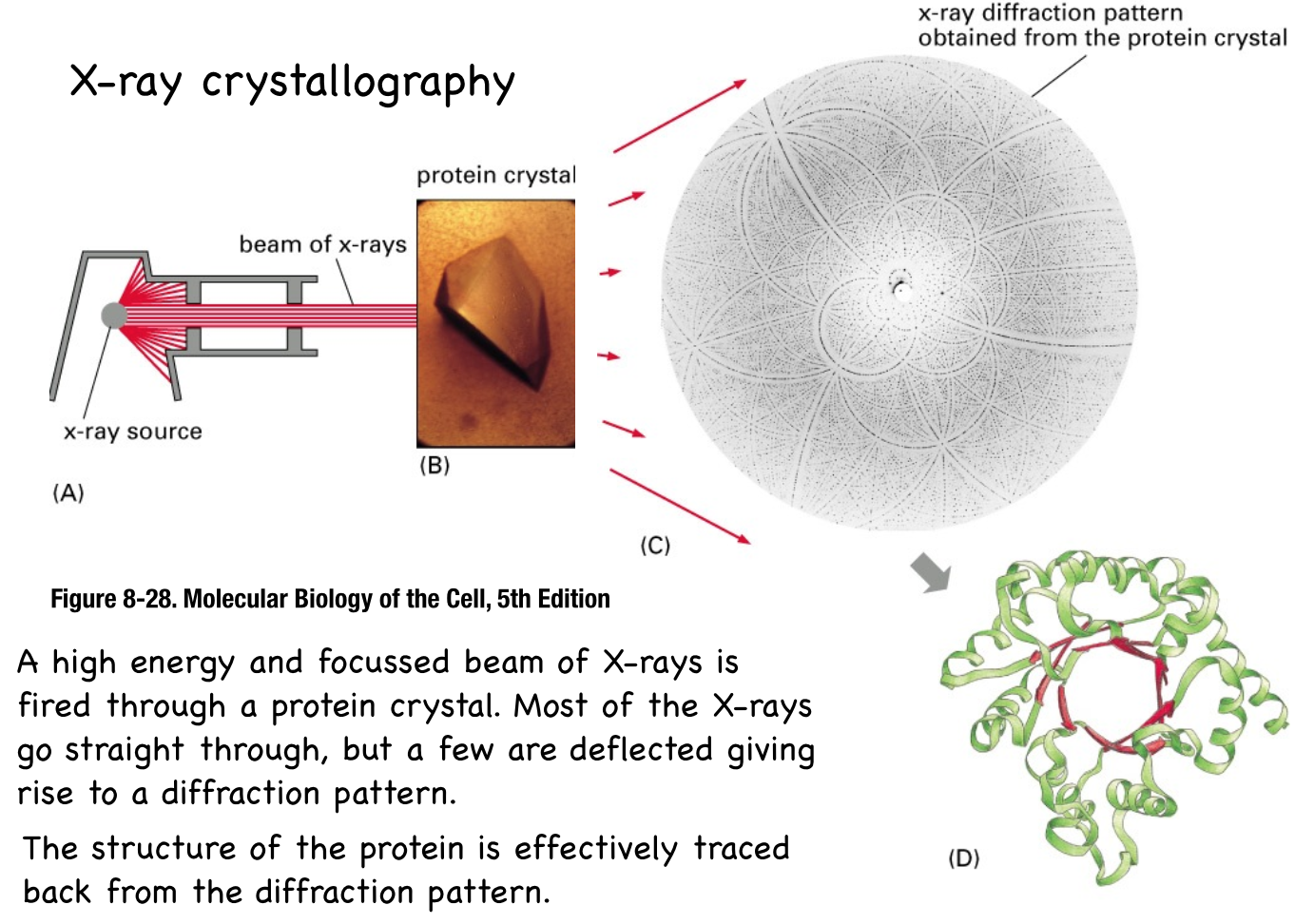
- X-ray crystallography
- NMR nuclear magnetic resonance
- In most atoms the ‘spin’ of
subatomic particles are paired
against each other, such that
the nucleus of the atom has no
overall spin.
- However, in some atoms (such as
1H,13C,15N) there are uneven
numbers of protons and neutrons so
the nucleus has a slight wobble in
the spin.
- Proteins are produced recombinantly, usually in
bacteria grown in media where the sole nutrient
source is 15N and/or 13C, so that all protein
produced are singly or doubly labelled with 15N
and/or 13C in every atom
- Proteins are produced recombinantly, usually in
bacteria grown in media where the sole nutrient
source is 15N and/or 13C, so that all protein
produced are singly or doubly labelled with 15N
and/or 13C in every atom
- ‘NMR active’ nuclei (like 1H or
13C) resonate at a specific
frequency in a strong magnetic
field.
- Depending on local
environment, different
protons resonate at slightly
different frequencies,
known as a chemical shift.
- NMR structure determination
is an iterative process, so one
arrives at several possible
structures which are usually
represented as an ensemble.
- In most atoms the ‘spin’ of
subatomic particles are paired
against each other, such that
the nucleus of the atom has no
overall spin.
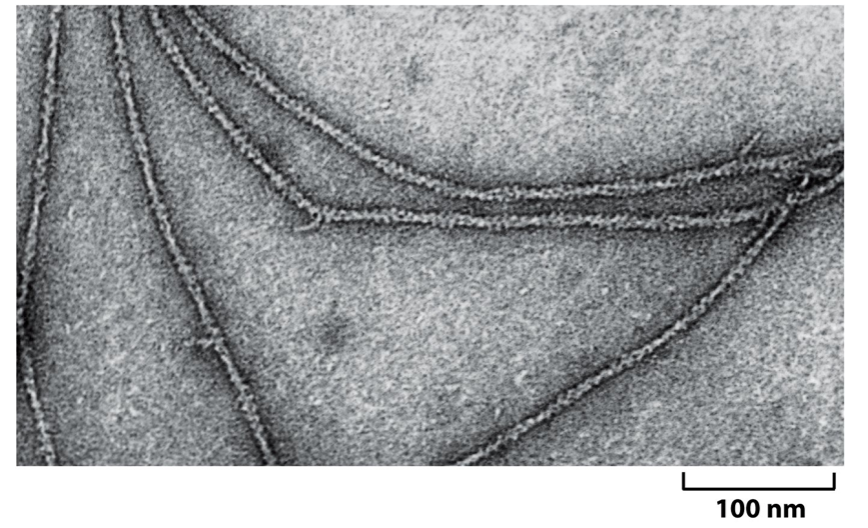
- Electron microscopy
- EM uses negative stain
(shown) or vitreous ice
(cryo-EM) to preserve the
specimen.
- Image analysis is then
employed to build up an average
structure.
- The more ordered and more
symmetrical the structure, the
easier the averaging process. e.g.
actin has helical symmetry,
viruses have radial symmetry
- The more ordered and more
symmetrical the structure, the
easier the averaging process. e.g.
actin has helical symmetry,
viruses have radial symmetry
- Image analysis is then
employed to build up an average
structure.
- EM uses negative stain
(shown) or vitreous ice
(cryo-EM) to preserve the
specimen.
- X-ray crystallography
Media attachments
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Want to create your own Mind Maps for free with GoConqr? Learn more.