6511226
Description

Mind Map by Greece Diaz, updated more than 1 year ago
|
|
Created by Greece Diaz
over 8 years ago
|
|
ENFERMEDADES DEGENERATIVAS
Y DESMIELINIZANTES
- Enfermedades desmielinizantes
- Son afecciones adquiridas que se caracterizan por el daño preferente de la mielina con conservación
relativa de los axones
- Déficit: perdida de mielina interfiere con la
transmisión de impulsos eléctricos a lo largo
de los axones
- HISTORIA NATURAL : se encuentra limitada por la capacidad del SNC de regenerar la mielina normal y por
el daño secundario de los axones que se desarrolla durante el proceso
- OTRAS ENFERMEDADES pueden afectar la mielina: leucoencefalopatía multifocal progresiva Y Enfermedad
de Pelizaeus-Merzbacher
- ¿Cómo se le llama a los trastornos
hereditarios que afectan la síntesis de
mielina y su recambio? leucodistrofias
- ¿Cómo se le llama a los trastornos
hereditarios que afectan la síntesis de
mielina y su recambio? leucodistrofias
- OTRAS ENFERMEDADES pueden afectar la mielina: leucoencefalopatía multifocal progresiva Y Enfermedad
de Pelizaeus-Merzbacher
- HISTORIA NATURAL : se encuentra limitada por la capacidad del SNC de regenerar la mielina normal y por
el daño secundario de los axones que se desarrolla durante el proceso
- Déficit: perdida de mielina interfiere con la
transmisión de impulsos eléctricos a lo largo
de los axones
- Son afecciones adquiridas que se caracterizan por el daño preferente de la mielina con conservación
relativa de los axones
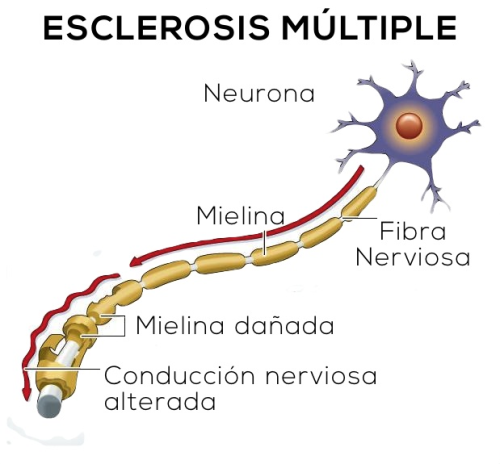
- ESCLEROSIS MÚLTIPLE
- Concepto: transtorno desmielinizante autoinmunitario se caracteriza por episodios bien delimitados y
separados en el tiempode defectos neurológicos atribuibles a lesiones de la sustancia blanca
- Frecuencia - - > más frecuente de los
transtornos con una prevalencia aprox. 1-1.000
- edad de presentación - - - >inicio en la infancia o
despues de los 50 años
- grupo etario más afectado - - > mujeres
- COMBINACION DE
FACTORES
GENETICOS Y
AMBIENTALES
- Haplotipo ampliado del complejo mayor de
histocompatibilidad DR2 - - > también puede haber
asociación con los genes receptores para IL-2 E IL7, genes
codificantes de citocinas y receptores, moléculas
coestimuladoras y de transmisión citoplásmaticas
- -- > inicia por Th17 y TH que reaccinan frente a los antígenos de mielina y que secretan citocinas
- - >TH secreta IFN7 que activa macrófagos y linfos y TH17 recluta neutrófilos -- -> se ha
propuesto que la autoinmunidad comienza por una infección vírica
- MICROSCOPIA - - - >
Enfermedad de la sustancia
blanca- - > cerebro y medula
espinal
- En placa activa se obseravan signos de
degradación + macrófagos abundantes
positivos en PAS
- Linfocitos +monocitos - - - > manguitis perivasculares en margen externo de la lesión Perdida de
oligodendrocitos - - -> las lesiones activas entan centradas sobre venas pequeñas
- Axones relativamente conservados en el seno de la placa y hay perdida de oligo Lesiones quiescentes - - >
cel. Inflamatorias desaparecen
- En el seno de las placas se observa poca/nada de mielina, nucleos de oligo reducidos y proliferación de
astrocitos con gliosis
- Los axones de placas glioticas antiguas demuestran diesmelirizacion -puede existir placas en sombras: no
hay delimitación neta entre sus. Gris afetctada/normal - - > suguiere remielinizacion fallida
- Los axones de placas glioticas antiguas demuestran diesmelirizacion -puede existir placas en sombras: no
hay delimitación neta entre sus. Gris afetctada/normal - - > suguiere remielinizacion fallida
- En el seno de las placas se observa poca/nada de mielina, nucleos de oligo reducidos y proliferación de
astrocitos con gliosis
- Axones relativamente conservados en el seno de la placa y hay perdida de oligo Lesiones quiescentes - - >
cel. Inflamatorias desaparecen
- Linfocitos +monocitos - - - > manguitis perivasculares en margen externo de la lesión Perdida de
oligodendrocitos - - -> las lesiones activas entan centradas sobre venas pequeñas
- En placa activa se obseravan signos de
degradación + macrófagos abundantes
positivos en PAS
- Haplotipo ampliado del complejo mayor de
histocompatibilidad DR2 - - > también puede haber
asociación con los genes receptores para IL-2 E IL7, genes
codificantes de citocinas y receptores, moléculas
coestimuladoras y de transmisión citoplásmaticas
- COMBINACION DE
FACTORES
GENETICOS Y
AMBIENTALES
- grupo etario más afectado - - > mujeres
- Macroscopicamente en fresco - - > esclerosis, que simula
placas pardo-griseasas de forma irregular y aspecto
vidrioso
- -areas de desmielizacion con bordes bien definidos
- -tamaño variable: pequeño o placas confluentes
- Placas localizadas en ventrículos laterales, nervios
ópticos y quiasma, encéfalo, tractos de vías
ascendentes, cerebelo y med, espinal--- alcanzar
sus.gris
- Placas localizadas en ventrículos laterales, nervios
ópticos y quiasma, encéfalo, tractos de vías
ascendentes, cerebelo y med, espinal--- alcanzar
sus.gris
- -tamaño variable: pequeño o placas confluentes
- -areas de desmielizacion con bordes bien definidos
- edad de presentación - - - >inicio en la infancia o
despues de los 50 años
- Frecuencia - - > más frecuente de los
transtornos con una prevalencia aprox. 1-1.000
- patogenia - - -> respuesta
autoinmunitaria frente a componentes de
la vaina mielinica
- Características clínicas - - > debilidad,
nistagmo, ataxia, oftalmoplejia
internuclear, lesiones sensitivas y
motoras: espasticidad u dificultad
control
- Alteración visual unilateral --- variante neuritis
óptica
- Alteración visual unilateral --- variante neuritis
óptica
- Características clínicas - - > debilidad,
nistagmo, ataxia, oftalmoplejia
internuclear, lesiones sensitivas y
motoras: espasticidad u dificultad
control
- Concepto: transtorno desmielinizante autoinmunitario se caracteriza por episodios bien delimitados y
separados en el tiempode defectos neurológicos atribuibles a lesiones de la sustancia blanca
- Enfermedades degenerativas
- Las enfermedades degenerativas son trastornos que se caracterizan por una pérdida progresiva de
neuronas, que afecta principalmente a grupos neuronales con ciertas funciones aunque no estén
adyacentes a estos.
- Se ven procesos patológicos comunes en enfermedades neurodegenerativas caracterizadas por cumulo de
agregados de proteínas por lo cual se agrupan en enfermedades llamadas proteinopatias
- las enfermedades neurodegenerativas tienen rasgos característicos como el cumulo de proteínas que son
resistentes a la degradación, generando respuestas de estrés con efecto toxico.
- Se produce una ganancia de función y perdida de función en caso de que la proteína sea toxica al tener un
cumulo excesivo y perdida de comunicación neuronal con la consecuente muerte o falta de respuesta
celular.
- Se produce una ganancia de función y perdida de función en caso de que la proteína sea toxica al tener un
cumulo excesivo y perdida de comunicación neuronal con la consecuente muerte o falta de respuesta
celular.
- las enfermedades neurodegenerativas tienen rasgos característicos como el cumulo de proteínas que son
resistentes a la degradación, generando respuestas de estrés con efecto toxico.
- Se ven procesos patológicos comunes en enfermedades neurodegenerativas caracterizadas por cumulo de
agregados de proteínas por lo cual se agrupan en enfermedades llamadas proteinopatias
- La principal causa de demencia en
ancianos es la enfermedad de
Alzheimer
- Se manifiesta por deterioro de inicio insidioso afectando la función intelectual superior. Cuadro clínico - - ->
deficiencia de memoria, en la orientación visoespacial, capacidad de juicio, la personalidad y el lenguaje.
- Después de 5 a 10 años el paciente termina con
discapacidad profunda, modo e inmóvil.
- Los pacientes desarrollan la enfermedad después de los 50
años de edad, aumentando la incidencia con la edad y su
prevalencia cada 5 años.
- Más del 40% de las personas con edad de 85-89 años de edad.
- Después de 5 a 10 años el paciente termina con
discapacidad profunda, modo e inmóvil.
- Su patología se desarrolla por acumulación de
dos proteínas (AB y tau) en regiones
específicas del cerebro, por una producción
excesiva y su eliminación defectuosa.
- Macroscópicamente
- Se observa atrofia de
diversos grados,
ensanchamiento de los
surcos cerebrales que
son mayores en lóbulos
frontales, temporal y
pariental.
- Se puede producir hidrocefalia ex
vacuo secundario a la reducción
del volumen cerebral Afecta
estructuras del lóbulo temporal
medial, hipocampo, corteza
entorrial y amígdala que afecta
precoz y estadios tardíos por
atrofia grave.
- Microscópicamente
- Se observan placas neuriticas (seniles) y ovillos neurofibrilares, con pérdida progresiva de neuronas que se
cubren de gliosis en dichas regiones de perdida.
- También se observa Angiopatía amiloidea cerebral, siendo el amieloide vascular es principalmente de tipo
AB.
- Otras causas de demencia son degenacion del lóbulo frontotemporal, demencia por cuerpos de Lewy,
degeneración corticobasal, degeneraciones espinocerebelosas.
- Otras causas de demencia son degenacion del lóbulo frontotemporal, demencia por cuerpos de Lewy,
degeneración corticobasal, degeneraciones espinocerebelosas.
- También se observa Angiopatía amiloidea cerebral, siendo el amieloide vascular es principalmente de tipo
AB.
- Se observan placas neuriticas (seniles) y ovillos neurofibrilares, con pérdida progresiva de neuronas que se
cubren de gliosis en dichas regiones de perdida.
- Microscópicamente
- Se puede producir hidrocefalia ex
vacuo secundario a la reducción
del volumen cerebral Afecta
estructuras del lóbulo temporal
medial, hipocampo, corteza
entorrial y amígdala que afecta
precoz y estadios tardíos por
atrofia grave.
- Se observa atrofia de
diversos grados,
ensanchamiento de los
surcos cerebrales que
son mayores en lóbulos
frontales, temporal y
pariental.
- Macroscópicamente
- Se manifiesta por deterioro de inicio insidioso afectando la función intelectual superior. Cuadro clínico - - ->
deficiencia de memoria, en la orientación visoespacial, capacidad de juicio, la personalidad y el lenguaje.
- Las enfermedades degenerativas son trastornos que se caracterizan por una pérdida progresiva de
neuronas, que afecta principalmente a grupos neuronales con ciertas funciones aunque no estén
adyacentes a estos.
- enfermedades degenerativas que afectan los ganglios basales y del tronco
- Se asocian con frecuencia a trastornos del movimiento,
incluyendo rigidez, posturas anormales y corea.
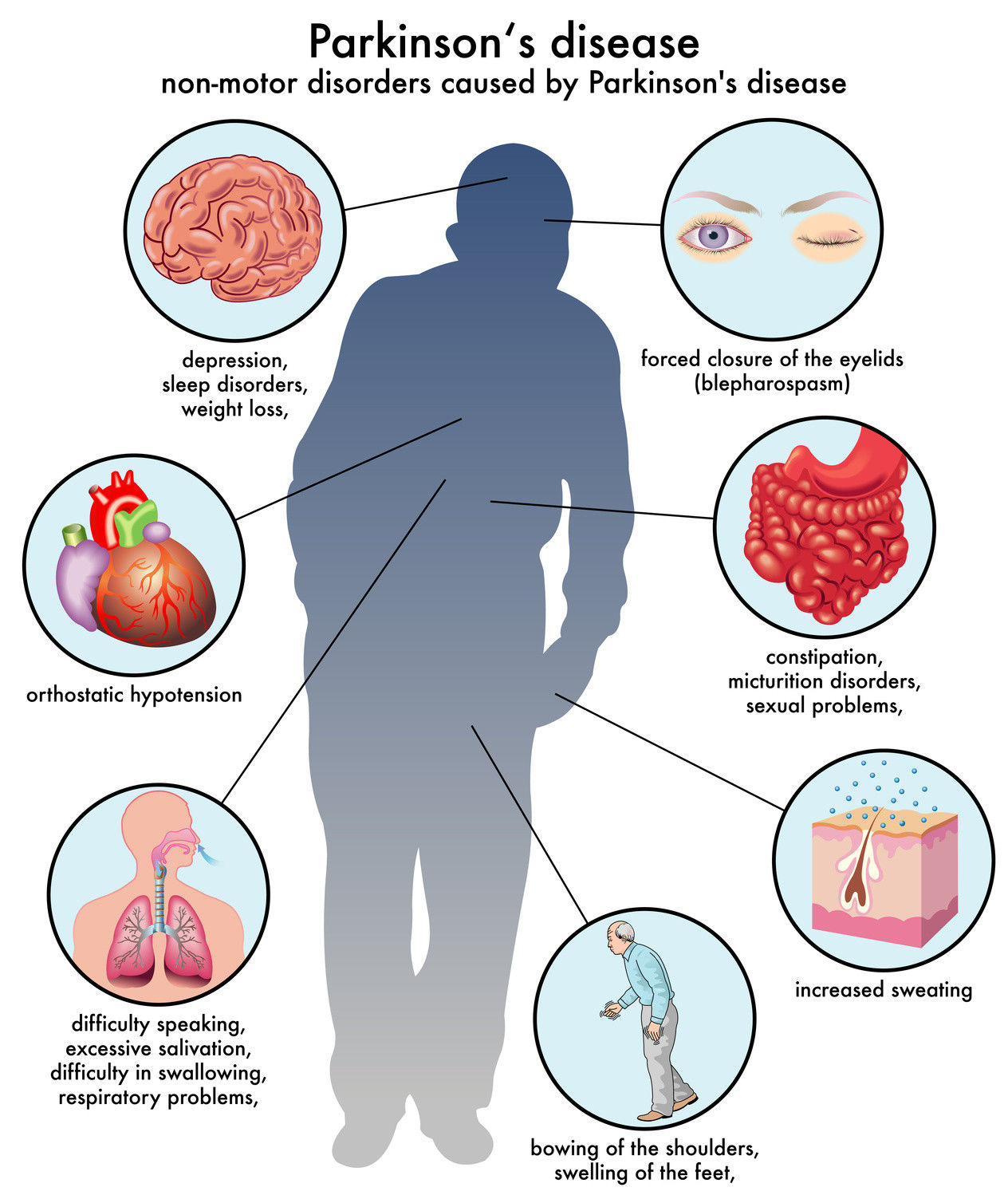
- Enfermedad de Parkinson
- Es un síndrome clínico que se caracteriza por la disminución de la expresión facial, lentitud de los
movimientos voluntarios, marcha festinante, postura inclinada, rigidez, temblor. Se observa en distintas
entidades patológicas que tienen en común el daño al sistema dopaminérgico nigroestriado.
- El diagnostico se puede basar en la
presencia de la tríada central del
parkinsonismo (temblor, rigidez y
bradicinesia).
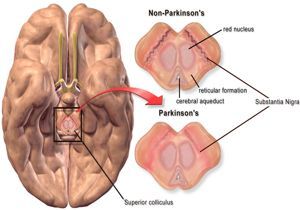
- PATOGENIA -----> Se asocia a la acumulación y agregación de las proteínas, alternaciones mitocondriales y perdida de
neuronas en la sustancia negra y otras regiones del cerebro.
- El diagnostico se puede basar en la
presencia de la tríada central del
parkinsonismo (temblor, rigidez y
bradicinesia).
- Morfología - - - -> Macroscopica: palidez de la sustancia ni gra y locus cerúleos.
- Microscópico: Perdida de las neuronas catecolaminérgicas pigmentadas, hay gliosis. Cuerpos de Lewy: son
inclusiones únicas o múltiples en el citoplasma de las neuronas que muestran una conformación de
filamentos finos, densamente empaquetados en el núcleo y más laxos en la zona periférica de la esfera
que rodea al núcleo. Son eosinofilicos, se ven redondeados o alargados y se observa un núcleo denso con
halo pálido.
- GENETICA
- Gen identificado como causa de la EP autosómica
dominante que codifica la alfa-sinucleína, se demostró que
esta proteína era un componente fundamental del cuerpo
de Lewy.
- Disfunción mitocondrial ha sido implicada como factor de la EP
en sus formas autosómicas recesivas, hay mutaciones en los
genes que codifican las proteínas DJ-1, PINK 1 y parkina.
- Mutaciones en el gen que codifica la LRRK2 son una causa más frecuente de EP autosómica dominante.
- Mutaciones en el gen que codifica la LRRK2 son una causa más frecuente de EP autosómica dominante.
- Disfunción mitocondrial ha sido implicada como factor de la EP
en sus formas autosómicas recesivas, hay mutaciones en los
genes que codifican las proteínas DJ-1, PINK 1 y parkina.
- Gen identificado como causa de la EP autosómica
dominante que codifica la alfa-sinucleína, se demostró que
esta proteína era un componente fundamental del cuerpo
de Lewy.
- GENETICA
- Microscópico: Perdida de las neuronas catecolaminérgicas pigmentadas, hay gliosis. Cuerpos de Lewy: son
inclusiones únicas o múltiples en el citoplasma de las neuronas que muestran una conformación de
filamentos finos, densamente empaquetados en el núcleo y más laxos en la zona periférica de la esfera
que rodea al núcleo. Son eosinofilicos, se ven redondeados o alargados y se observa un núcleo denso con
halo pálido.
- Es un síndrome clínico que se caracteriza por la disminución de la expresión facial, lentitud de los
movimientos voluntarios, marcha festinante, postura inclinada, rigidez, temblor. Se observa en distintas
entidades patológicas que tienen en común el daño al sistema dopaminérgico nigroestriado.
- Se asocian con frecuencia a trastornos del movimiento,
incluyendo rigidez, posturas anormales y corea.
- HERNANDEZ DIAZ DE LEON GRECIA
Media attachments
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Want to create your own Mind Maps for free with GoConqr? Learn more.