6495940
Question 1
Question
[blank_start]Tumorigenesis[blank_end] is the result of an accumulation of key [blank_start]mutations[blank_end] in [blank_start]cancer-critical[blank_end] genes in a cell’s progeny, which eventually causes seriously [blank_start]dysregulated[blank_end] cell growth and survival. Exposure to environmental [blank_start]mutagens[blank_end], failure of DNA [blank_start]repair mechanisms[blank_end] and [blank_start]inherited mutations[blank_end] contribute to the accumulation of [blank_start]genetic lesions[blank_end].
Answer
-
Tumorigenesis
-
mutations
-
cancer-critical
-
dysregulated
-
mutagens
-
repair mechanisms
-
inherited mutations
-
genetic lesions
Question 2
Question
Genes whose mutated version leads to [blank_start]upregulation[blank_end] in the protein product’s activity or in its expression in tumours (usually caused by a [blank_start]gain-of-function[blank_end] mutation) are called [blank_start]oncogenes[blank_end]. In contrast, those genes whose products are [blank_start]downregulated[blank_end] in tumours (either affecting their activity or expression), usually by [blank_start]loss-of-function[blank_end] mutations, are called [blank_start]tumour suppressor[blank_end] genes.
Answer
-
upregulation
-
gain-of-function
-
oncogenes
-
downregulated
-
loss-of-function
-
tumour suppressor
Question 3
Question
The initial [blank_start]random genetic lesion[blank_end] occurs within a normal cell, leading to the clinical development of a [blank_start]benign hyperplasia[blank_end]. This progresses with further mutations to [blank_start]dysplasia[blank_end], through to fully [blank_start]malignant cancer[blank_end], which is [blank_start]invasive[blank_end] and has [blank_start]metastatic potential[blank_end]. Although [blank_start]clonal[blank_end] in origin, tumours acquire [blank_start]genetic instability[blank_end] and contain [blank_start]heterogeneous clones[blank_end].
Answer
-
random genetic lesion
-
benign hyperplasia
-
dysplasia
-
malignant cancer
-
invasive
-
metastatic potential
-
clonal
-
genetic instability
-
heterogeneous clones
Question 4
Question
In medical terms, tumours are traditionally classified according to their [blank_start]tissue of origin[blank_end]. The majority of cancers in humans are [blank_start]carcinomas[blank_end], from epithelial cells. This is because they have a [blank_start]rapid turnover rate[blank_end], which increases the likelihood of mutations occurring. Moreover, they are often exposed to chemical and physical [blank_start]mutagens[blank_end], as they are the first barrier that environmental agents encounter in the body. [blank_start]Gut, lung and skin epithelia[blank_end] are common sites for tumours for this reason.
Other types of cancers are [blank_start]sarcomas[blank_end], from muscle or connective tissue, or [blank_start]leukaemias[blank_end], from haemopoietic cells.
Answer
-
tissue of origin
-
carcinomas
-
rapid turnover rate
-
mutagens
-
Gut, lung and skin epithelia
-
sarcomas
-
leukaemias
Question 5
Question
The key [blank_start]tumorigenic[blank_end] properties that cells acquire through successive rounds of mutation include: [blank_start]genetic instability[blank_end], self-sufficiency in growth signals (usually by activating [blank_start]oncogenes[blank_end]), insensitivity to growth-inhibitory signals (often involving inactivation of [blank_start]tumour suppressor genes[blank_end]), avoidance of [blank_start]apoptosis[blank_end] and replicative [blank_start]senescence[blank_end], sustained [blank_start]angiogenesis[blank_end], and tissue invasion and [blank_start]metastasis[blank_end].
Answer
-
tumorigenic
-
genetic instability
-
oncogenes
-
tumour suppressor genes
-
apoptosis
-
senescence
-
angiogenesis
-
metastasis
Question 6
Question
Genetic [blank_start]instability[blank_end] accelerates the rate of accumulation of key mutations in [blank_start]cancer-critical[blank_end] genes.
Genetic instability can be a result of [blank_start]mutation[blank_end] in any of the many genes whose products check, detect and repair [blank_start]damaged[blank_end] DNA.
Cancer cells show [blank_start]increased[blank_end] mutation rates compared to normal cells, due to genetic instability.
Answer
-
instability
-
cancer-critical
-
mutation
-
damaged
-
increased
-
decreased
Question 7
Question
The type of genetic [blank_start]instability[blank_end] within a cell reflects the [blank_start]pathway[blank_end] that has been disrupted, e.g. loss of [blank_start]mismatch repair genes[blank_end] leads to a superficially [blank_start]normal karyotype[blank_end], with many [blank_start]point mutations[blank_end] and short repeats, while loss of [blank_start]ATM or p53[blank_end] leads to [blank_start]translocations[blank_end] resulting from [blank_start]double-strand DNA breaks[blank_end].
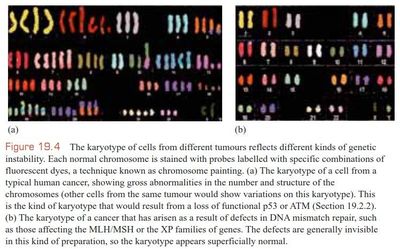{kind=link}
Answer
-
instability
-
pathway
-
mismatch repair genes
-
normal karyotype
-
point mutations
-
ATM or p53
-
translocations
-
double-strand DNA breaks
Question 8
Question
The [blank_start]p53[blank_end] protein normally triggers [blank_start]cell cycle arrest[blank_end] in response to [blank_start]DNA damage[blank_end], leading to DNA [blank_start]repair[blank_end] or [blank_start]apoptosis[blank_end] if damage is severe. Continued [blank_start]replication[blank_end] in p53-deficient cells leads to increasing [blank_start]genetic damage[blank_end] and [blank_start]chromosomal[blank_end] instability.
50% of [blank_start]tumours[blank_end] have mutations in the p53 gene.
Answer
-
p53
-
cell cycle arrest
-
DNA damage
-
repair
-
apoptosis
-
replication
-
genetic damage
-
chromosomal
-
tumours
Question 9
Question
[blank_start]Radiotherapy[blank_end] and [blank_start]chemotherapeutic[blank_end] drugs rely on [blank_start]damaging[blank_end] the DNA of [blank_start]tumour cells[blank_end] so severely that they undergo [blank_start]mitotic failure[blank_end] and die, whilst normal cells, with normal [blank_start]repair mechanisms[blank_end], either undergo [blank_start]apoptosis[blank_end] or [blank_start]repair the damage[blank_end] and survive.
Answer
-
Radiotherapy
-
chemotherapeutic
-
damaging
-
tumour cells
-
mitotic failure
-
repair mechanisms
-
apoptosis
-
repair the damage
Question 10
Question
Label the diagram of proto-oncogenes becoming oncogenes
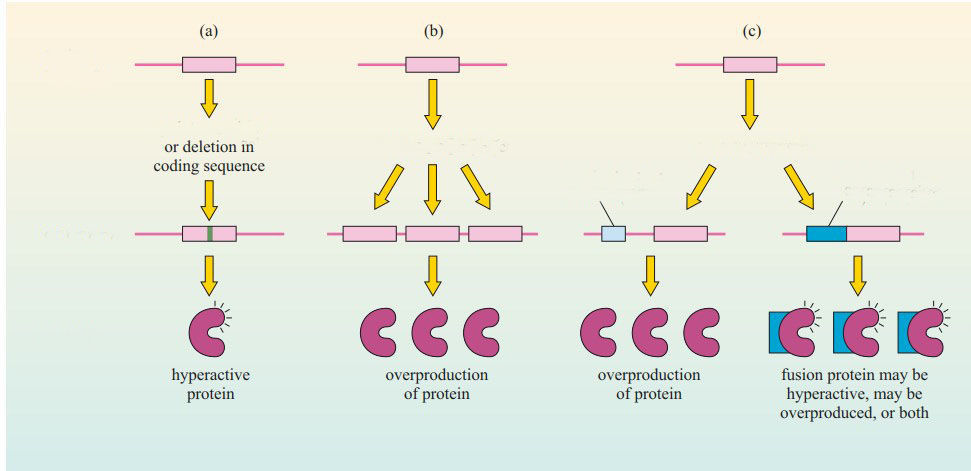{kind=link}
Answer
-
proto-oncogene
-
oncogene
-
protein
-
point mutation
-
gene amplification
-
rearrangements
-
new promoter region
-
fusion to another gene
Question 11
Question
[blank_start]Oncogenes[blank_end] can be identified by their ability to transform [blank_start]immortalized fibroblast[blank_end] cell lines and have often been discovered in [blank_start]viruses[blank_end]. Only a [blank_start]single copy[blank_end] needs to be mutated, because the effect is [blank_start]dominant[blank_end]. [blank_start]Point[blank_end] mutations, [blank_start]deletions[blank_end], amplification and chromosome and gene [blank_start]rearrangement[blank_end] can all produce [blank_start]gain-of-function[blank_end] effects.
Answer
-
Oncogenes
-
immortalized fibroblast
-
viruses
-
single copy
-
dominant
-
Point
-
deletions
-
rearrangement
-
gain-of-function
Question 12
Question
Self-sufficiency in [blank_start]growth signals[blank_end] can arise through [blank_start]activation[blank_end] of individual components of a [blank_start]growth-promoting signalling[blank_end] pathway. Proteins encoded by [blank_start]proto-oncogenes[blank_end] include [blank_start]growth factors[blank_end] (e.g. PDGF), [blank_start]receptors[blank_end] (EGF receptors), cytoplasmic [blank_start]signalling molecules[blank_end] (Ras, Src and Bcr–Abl), and [blank_start]transcription[blank_end] factors (Myc).
Answer
-
growth signals
-
activation
-
growth-promoting signalling
-
proto-oncogenes
-
growth factors
-
receptors
-
signalling molecules
-
transcription
Question 13
Question
[blank_start]Tumour suppressor genes[blank_end], such as [blank_start]Rb[blank_end] , were originally discovered through study of rare [blank_start]inherited[blank_end] cancers. For cell tumorigenesis to occur, [blank_start]both copies[blank_end] need to be inactivated (e.g. by point [blank_start]mutation[blank_end], deletion, [blank_start]methylation[blank_end]), which is [blank_start]less likely[blank_end] to occur than activation of a [blank_start]single oncogene allele[blank_end], but has very [blank_start]profound effects[blank_end] on the cell.
Answer
-
Tumour suppressor genes
-
Rb
-
inherited
-
both copies
-
mutation
-
methylation
-
less likely
-
single oncogene allele
-
profound effects
Question 14
Question
Loss of [blank_start]tumour suppressor genes[blank_end] can cause [blank_start]insensitivity[blank_end] to either [blank_start]external growth-inhibitory[blank_end] signals, or to [blank_start]internal checkpoints[blank_end]. Loss of [blank_start]functional Rb[blank_end] causes deregulation of the [blank_start]G1/S checkpoint[blank_end]. Loss of [blank_start]p53[blank_end] allows cells with sustained [blank_start]DNA damage[blank_end] to continue to [blank_start]replicate[blank_end], when normally they would be [blank_start]arrested[blank_end] at entry into [blank_start]S phase[blank_end], by p53/p21CIP-mediated mechanisms, until the damage was [blank_start]repaired[blank_end].
Answer
-
tumour suppressor genes
-
insensitivity
-
external growth-inhibitory
-
internal checkpoints
-
functional Rb
-
G1/S checkpoint
-
p53
-
DNA damage
-
replicate
-
arrested
-
S phase
-
repaired
Question 15
Question
[blank_start]Dysregulation[blank_end] of the pathways of [blank_start]apoptosis[blank_end] is as important in the microevolution of [blank_start]tumours[blank_end] as the changes that increase the rate of cell [blank_start]proliferation[blank_end]. Tumour cells acquire mutations that cause them to resist [blank_start]apoptotic signals[blank_end] by either [blank_start]inhibiting[blank_end] pro-apoptotic signals or [blank_start]activating[blank_end] anti-apoptotic pathways. Both [blank_start]intrinsic[blank_end] and extrinsic pathways can be [blank_start]disrupted[blank_end] in tumorigenic cells.
Answer
-
Dysregulation
-
apoptosis
-
tumours
-
proliferation
-
apoptotic signals
-
inhibiting
-
activating
-
intrinsic
-
disrupted
Question 16
Question
Tumour cells acquire mutations that cause them to avoid [blank_start]replicative senescence[blank_end].
One mechanism involves [blank_start]p53 inactivation[blank_end], so that cells with damaged [blank_start]telomeres[blank_end] continue to divide. Tumour cells may also reactivate [blank_start]telomerase[blank_end], often after significant [blank_start]chromosomal[blank_end] damage has been sustained, thus allowing clonal tumour cells that would otherwise be [blank_start]non-viable[blank_end], to survive and proliferate.
Answer
-
replicative senescence
-
p53 inactivation
-
telomeres
-
telomerase
-
chromosomal
-
non-viable
Question 17
Question
Most cells suffering from [blank_start]hypoxia[blank_end] will synthesize and secrete [blank_start]vascular endothelial growth factor[blank_end] (VEGF) which promotes [blank_start]angiogenesis[blank_end] by attracting [blank_start]endothelial cells[blank_end] which then form new [blank_start]capillaries[blank_end].
Tumour cells in solid tumours do this too.
Answer
-
hypoxia
-
vascular endothelial growth factor
-
angiogenesis
-
endothelial cells
-
capillaries
Question 18
Question
Solid [blank_start]tumours[blank_end] secrete [blank_start]VEGF[blank_end] and other [blank_start]angiogenic[blank_end] factors which induce [blank_start]angiogenesis[blank_end].
Answer
-
tumours
-
VEGF
-
angiogenic
-
angiogenesis
Question 19
Question
Tissue [blank_start]invasion[blank_end] by tumours requires the breakdown of normal [blank_start]cell–ECM[blank_end] and cell–cell [blank_start]adhesion[blank_end] interactions; for example, [blank_start]E-cadherin[blank_end] is absent in some tumours.
Answer
-
invasion
-
cell–ECM
-
adhesion
-
E-cadherin
Question 20
Question
[blank_start]Metastasis[blank_end] is very inefficient and poorly understood. The cells need to break through the [blank_start]basal lamina[blank_end] of blood or lymphatic vessels, probably aided by the secretion of serine proteases or [blank_start]matrix metalloproteases[blank_end]. Once in the circulation, they lodge in [blank_start]capillary beds[blank_end] (or if in lymphatics, lodge in draining lymph nodes) and may then colonize the new tissue. Successful [blank_start]colonization[blank_end] probably depends on the degree of [blank_start]self-sufficiency[blank_end] of the [blank_start]metastatic[blank_end] cells.
Answer
-
Metastasis
-
basal lamina
-
matrix metalloproteases
-
capillary beds
-
colonization
-
self-sufficiency
-
metastatic
Question 21
Question
Colorectal cancer is a suitable model for the study of [blank_start]tumour progression[blank_end], due to the availability of surgically removed early polyps. There are characteristic [blank_start]genetic lesions[blank_end], some relating to the [blank_start]specific cell type[blank_end], and some tending to occur [blank_start]earlier[blank_end] or later in the progression.
Answer
-
tumour progression
-
genetic lesions
-
specific cell type
-
earlier
Question 22
Question
Mutations in the Wnt pathway genes ( APC and β -catenin ), which maintain [blank_start]gut epithelial stem cells[blank_end], are common in human colorectal cancer; [blank_start]APC gene mutations[blank_end] are found in patients with familial adenomatous polyposis coli.
Answer
-
gut epithelial stem cells
-
APC gene mutations
Question 23
Question
Mutations of [blank_start]p53 and K - ras[blank_end] are particularly common in human colorectal cancer, while mutations in genes encoding proteins of the [blank_start]TGF pathway[blank_end] ( Smad4, TGFrII ), in DNA [blank_start]mismatch[blank_end] repair genes and in [blank_start]tyrosine phosphatase[blank_end] genes are found at lower frequencies.
Answer
-
p53 and K - ras
-
TGF pathway
-
mismatch
-
tyrosine phosphatase
Question 24
Question
[blank_start]p53[blank_end] is involved in activation of the [blank_start]G1/S cell cycle[blank_end] checkpoint in response to [blank_start]DNA damage[blank_end], which leads to cell cycle [blank_start]arrest[blank_end]. It also acts as an inducer of downstream pathways of [blank_start]DNA repair[blank_end] and (if repair is unsuccessful) [blank_start]apoptosis[blank_end], and is involved in the induction of [blank_start]replicative senescence[blank_end]
Answer
-
p53
-
G1/S cell cycle
-
DNA damage
-
arrest
-
DNA repair
-
apoptosis
-
replicative senescence
Want to create your own Quizzes for free with GoConqr? Learn more.