23524983
Beschreibung
Mindmap von Sarah Novaes, aktualisiert more than 1 year ago
|
|
Erstellt von Sarah Novaes
vor mehr als 4 Jahre
|
|
Imunodeficiências
Primárias
- São distúrbios determinados geneticamente, de modo a causar
um aumento na suscetibilidade a infecções. Podem ocorrer de
maneira isolada, ou associados a alguma síndrome.
- Manifestam-se precocemente em bebês e crianças, surgindo
desde o seu nascimento. Podem ser assintomáticas ou
altamente letais no primeiro ano de vida.
- As imunodeficiências primárias (ou congênitas) podem ser divididas
em 4 tipos principais, de acordo com o componente imunológico
deficiente:
- Imunodeficiência Humoral
- É o tipo de imunodeficiência primária mais comum
na população humana.
- São doenças relacionadas a defeitos na célula B que causam
deficiência de anticorpos, predispondo o paciente a desenvolver
infecções bacterianas.
- Alguns exemplos dessas doenças são: Agamaglobulinemia, Deficiência
Seletiva de IgA, Síndrome da Hiper-IgM, Hipogamaglobulinemia
Transitória da Infância, dentre outras. As duas primeiras são as mais
comuns.
- AGAMAGLOBULINEMIA
- É mais comum em crianças do sexo
masculino, manifestando-se entre 5 e 9
meses após o nascimento
- As imunoglobulinas de origem maternas são substituidas pelas da
criança, gerando assim uma deficiência no sistema imunológico da
mesma.
- O defeito apresentado nesta doença é a ausência ou mutação de gene responsável por originar a
tirosina quinase (responsáveis pela fosforilação de substratos proteicos), essencial no processo de
amadurecimento.
- O linfócito B será prejudicado, uma vez que
não poderá atingir seu estado de maturação
- Algumas das consequencias apresentadas pelo paciente são:
- Diminuição do número de
células B e anticorpos
- Ausência de plasmócitos
- Diminuição do número de células T
- Surgimento de infecções
recorrentes, como: pneumonia,
meningite, otite e etc.
- Diminuição do número de
células B e anticorpos
- Algumas das consequencias apresentadas pelo paciente são:
- O linfócito B será prejudicado, uma vez que
não poderá atingir seu estado de maturação
- O defeito apresentado nesta doença é a ausência ou mutação de gene responsável por originar a
tirosina quinase (responsáveis pela fosforilação de substratos proteicos), essencial no processo de
amadurecimento.
- As imunoglobulinas de origem maternas são substituidas pelas da
criança, gerando assim uma deficiência no sistema imunológico da
mesma.
- É mais comum em crianças do sexo
masculino, manifestando-se entre 5 e 9
meses após o nascimento
- DEFICIÊNCIA SELETIVA DE IgA
- A deficiência seletiva da imunoglobulina A consiste em níveis
de IgA < 7mg/dL, onde o paciente apresenta quantidades
normais de IgG e IgM.
- É a imunodeficiência primária mais comum, de modo que alguns pacientes podem ser
assintomáticos, enquanto outros apresentam infecções recorrentes, e desenvolvimento de
doenças autoimunes (ex: LES).
- O IgA é a principal imunoglobulina presente nas mucosas. Sua ausência, portanto,
tende a predispor o paciente a desenvolver infecções bacterianas e virais,
principalmente nas vias aéreas superiores, e sistemas gastrointestinal e urogenital.
- Fármacos como fenitoína, sulfassalazina, ouro e d-penicilamina podem levar à
deficiência de IgA em alguns pacientes.
- Fármacos como fenitoína, sulfassalazina, ouro e d-penicilamina podem levar à
deficiência de IgA em alguns pacientes.
- O IgA é a principal imunoglobulina presente nas mucosas. Sua ausência, portanto,
tende a predispor o paciente a desenvolver infecções bacterianas e virais,
principalmente nas vias aéreas superiores, e sistemas gastrointestinal e urogenital.
- Os linfócitos B secretam incialmente IgM, e após
interagirem com as células T auxiliares, serão formados (a
depender da citocina produzida) IgG, IgA ou IgE.
- O defeito apresentado por pacientes portadores desta condição,
consiste no bloqueio da diferenciação de células B em plasmócitos
secretores de IgA.
- O defeito apresentado por pacientes portadores desta condição,
consiste no bloqueio da diferenciação de células B em plasmócitos
secretores de IgA.
- É a imunodeficiência primária mais comum, de modo que alguns pacientes podem ser
assintomáticos, enquanto outros apresentam infecções recorrentes, e desenvolvimento de
doenças autoimunes (ex: LES).
- A deficiência seletiva da imunoglobulina A consiste em níveis
de IgA < 7mg/dL, onde o paciente apresenta quantidades
normais de IgG e IgM.
- AGAMAGLOBULINEMIA
- Alguns exemplos dessas doenças são: Agamaglobulinemia, Deficiência
Seletiva de IgA, Síndrome da Hiper-IgM, Hipogamaglobulinemia
Transitória da Infância, dentre outras. As duas primeiras são as mais
comuns.
- São doenças relacionadas a defeitos na célula B que causam
deficiência de anticorpos, predispondo o paciente a desenvolver
infecções bacterianas.
- Quando suspeitar?
- Início tardio dos sintomas, já que nas
primeiras décadas de vida é comum que o
paciente seja assintomático.
- Infecções recorrentes
no trato respiratório
- Respostas positivas ao uso de
antibióticos
- Diarréia
- Atrofia de orgãos
linfóides periféricos
- Início tardio dos sintomas, já que nas
primeiras décadas de vida é comum que o
paciente seja assintomático.
- É o tipo de imunodeficiência primária mais comum
na população humana.
- Imunodeficiência Celular ou Combinada
- É um tipo de imunodeficiência grave, que envolve defeitos no
desenvolvimento dos linfócitos T, podendo ser associados ou não
as células B.
- São responsáveis por cerca de 20% das
imunodeficiências primárias
- Em algumas formas de imunodeficiência combinada, os níveis de imunoglobulinas são
normais ou elevados, devido a atividade deficiente da célula T. Logo, ocorre diminuição da
formação de anticorpos.
- Alguns exemplos dessas imunodeficiências são: Imunodeficiência Combinada Severa (SCID -
principal tipo), Deficiência em adenosina deamidase (ADA), e Síndrome do Linfócito Nu.
- IMUNODEFICIÊNCIA COMBINADA SEVERA (SCID)
- É uma doença potencialmente fatal, presente desde o
nascimento. Pode ser causada por mutações em diversos
genes, sendo todas as formas de caráter hereditário.
- Caracteriza-se pela ausência ou redução de linfócitos T
- A forma mais comum resulta de uma mutação em um gene do cromossomo X,
ocorrendo quase exclusivamente em indivíduos do sexo masculino. A mutação
atinge receptores das citocinas IL-2 (ativador de células T), IL-15 (ativador de
células NK), IL-7 (maturação de células línfoides e mieloides).
- Amadurecimento prejudicado dos linfócitos T e células NK
- Indivíduos com imunodeficiência combinada grave, o sistema imunológico não fornece, praticamente,
nenhuma proteção contra bactérias, vírus e fungos. Resultando em infecções repetidas e persistentes.
Também podem desenvolver candidíase, diarréia, e em casos mais graves, pneumonias.
- Devido a isso, as crianças não crescem nem se desenvolvem
normalmente ( condição conhecida como insuficiência de
desenvolvimento geral).
- Devido a isso, as crianças não crescem nem se desenvolvem
normalmente ( condição conhecida como insuficiência de
desenvolvimento geral).
- Indivíduos com imunodeficiência combinada grave, o sistema imunológico não fornece, praticamente,
nenhuma proteção contra bactérias, vírus e fungos. Resultando em infecções repetidas e persistentes.
Também podem desenvolver candidíase, diarréia, e em casos mais graves, pneumonias.
- Amadurecimento prejudicado dos linfócitos T e células NK
- A forma mais comum resulta de uma mutação em um gene do cromossomo X,
ocorrendo quase exclusivamente em indivíduos do sexo masculino. A mutação
atinge receptores das citocinas IL-2 (ativador de células T), IL-15 (ativador de
células NK), IL-7 (maturação de células línfoides e mieloides).
- Caracteriza-se pela ausência ou redução de linfócitos T
- É uma doença potencialmente fatal, presente desde o
nascimento. Pode ser causada por mutações em diversos
genes, sendo todas as formas de caráter hereditário.
- IMUNODEFICIÊNCIA COMBINADA SEVERA (SCID)
- Alguns exemplos dessas imunodeficiências são: Imunodeficiência Combinada Severa (SCID -
principal tipo), Deficiência em adenosina deamidase (ADA), e Síndrome do Linfócito Nu.
- Em algumas formas de imunodeficiência combinada, os níveis de imunoglobulinas são
normais ou elevados, devido a atividade deficiente da célula T. Logo, ocorre diminuição da
formação de anticorpos.
- São responsáveis por cerca de 20% das
imunodeficiências primárias
- Quando suspeitar?
- Início precoce dos sintomas
- Infecções graves
- Resposta negativa a antibióticos
- Defeitos no crescimento
- Desnutrição
- Diarréia crônica
- Doença inflamatória cutânea
- Ausência de orgãos linfóides
- Início precoce dos sintomas
- É um tipo de imunodeficiência grave, que envolve defeitos no
desenvolvimento dos linfócitos T, podendo ser associados ou não
as células B.
- Deficiência de Fagócitos
- Os defeitos de células fagocíticas representam cerca de 10
a 15% das imunodeficiências primárias e comprometem a
capacidade fagocitária de destruir patógenos
- Alguns exemplos de doenças relacionadas a esse tipo são: Doença Granulomatosa Crônica (principal
exemplo), Deficiência de Adesão Leucocitária (LAD), Neutropenia Congênita Grave e Neutropenia
Cíclica.
- DOENÇA GRANULOMATOSA CRÔNICA
- Caracteriza-se por leucócitos incapazes de produzir compostos ativos de
oxigênio, além de defeitos na função microbicida das células fagocíticas.
- Cerca de 50% dos casos da doença são relacionados a caracteristicas recessivas
associadas ao cromossomo X, sendo assim, mais frequente em indivíduos do
sexo masculino.
- Portanto, a falha na atividade microbicida dos fagócitos ocorre devido a uma
mutação do complexo enzimático responsável pela produção de produtos tóxicos
derivados do oxigênio.
- Sendo assim, indivíduos que herdam essa falha são acometidos de infecções
recorrentes de origem fúngica e bacteriana, formando também granulomas.
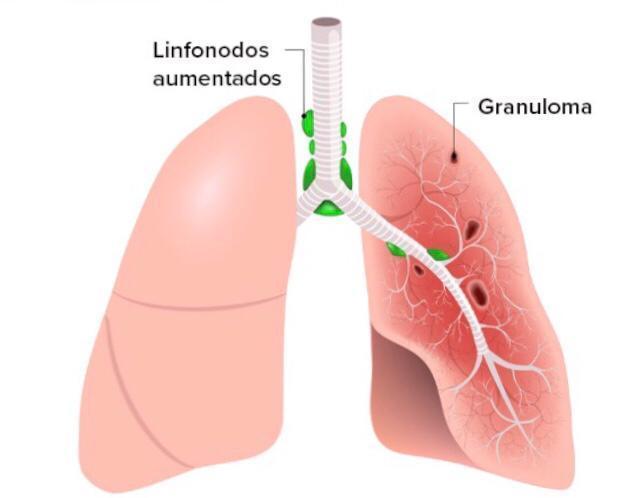
- Lesões granulomatosas múltiplas ocorrem nos pulmões, fígado, linfonodos e tratos
gastrointestinal e geniturinário (causando obstrução). Também pode ocorrer atraso no
crescimento.
- Lesões granulomatosas múltiplas ocorrem nos pulmões, fígado, linfonodos e tratos
gastrointestinal e geniturinário (causando obstrução). Também pode ocorrer atraso no
crescimento.
- Sendo assim, indivíduos que herdam essa falha são acometidos de infecções
recorrentes de origem fúngica e bacteriana, formando também granulomas.
- Portanto, a falha na atividade microbicida dos fagócitos ocorre devido a uma
mutação do complexo enzimático responsável pela produção de produtos tóxicos
derivados do oxigênio.
- Cerca de 50% dos casos da doença são relacionados a caracteristicas recessivas
associadas ao cromossomo X, sendo assim, mais frequente em indivíduos do
sexo masculino.
- Caracteriza-se por leucócitos incapazes de produzir compostos ativos de
oxigênio, além de defeitos na função microbicida das células fagocíticas.
- DOENÇA GRANULOMATOSA CRÔNICA
- Alguns exemplos de doenças relacionadas a esse tipo são: Doença Granulomatosa Crônica (principal
exemplo), Deficiência de Adesão Leucocitária (LAD), Neutropenia Congênita Grave e Neutropenia
Cíclica.
- Quando suspeitar?
- Início precoce
- Deficiência ponderal e estrutural
- Abcessos recorrentes
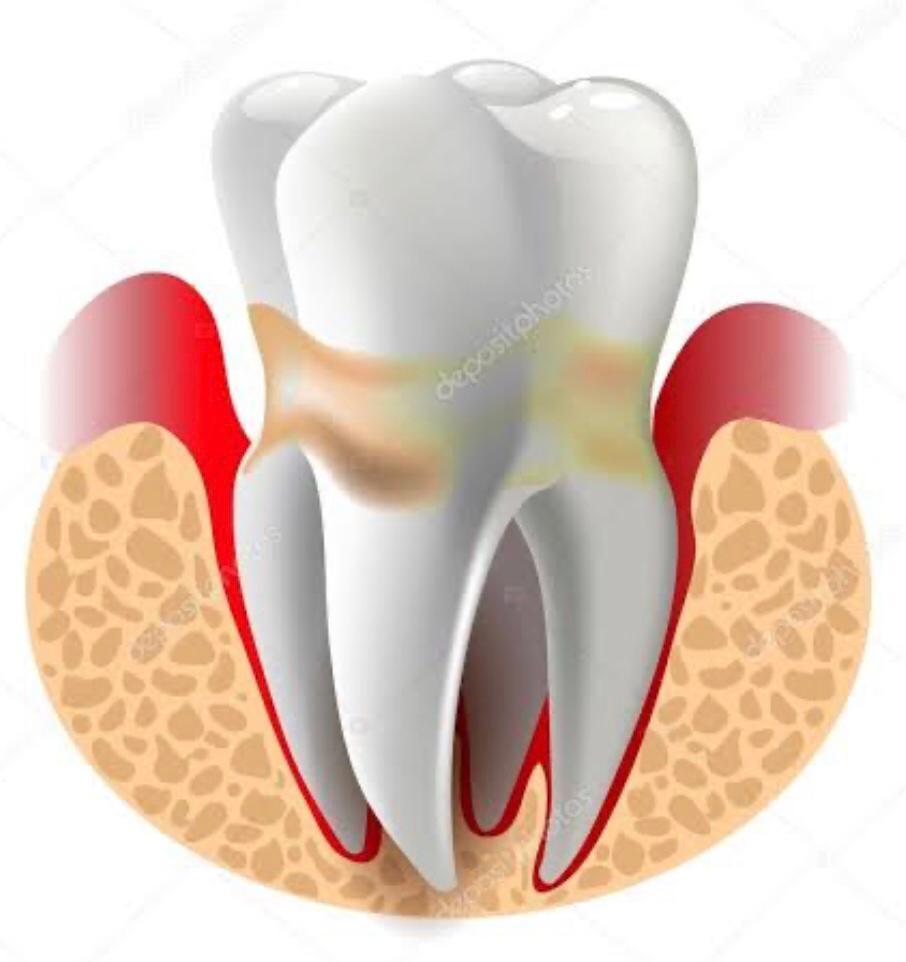
- Estomatite e periodontite
- Atraso na queda do
cordão umbilical
- Infecções recorrentes
- Início precoce
- Os defeitos de células fagocíticas representam cerca de 10
a 15% das imunodeficiências primárias e comprometem a
capacidade fagocitária de destruir patógenos
- Deficiência de Complemento
- As deficiências do sistema complemento são as mais raras,
englobando cerca de 2% dos casos.
- São distúrbios isolados de componentes ou os inibidores do
complemento, podendo ser hereditários ou adquiridos
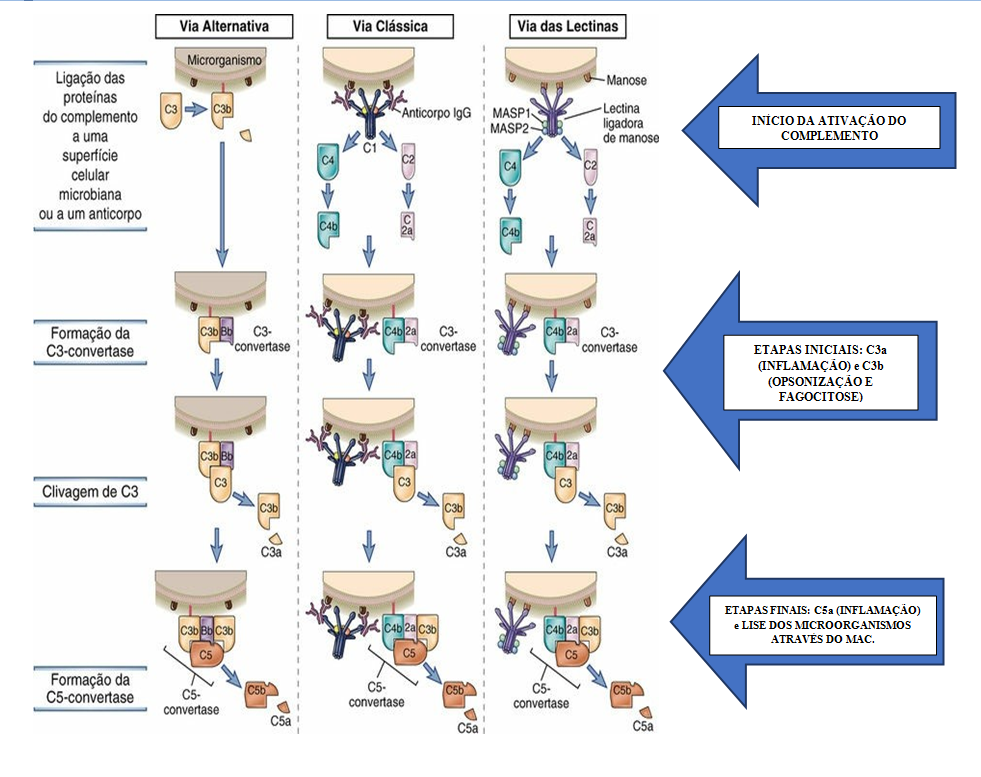
- O sistema complemento é um sistema de proteínas que
possui 3 vias de ativação: clássica, alternativa e das lectinas.
- As deficiências hereditárias são autossômicas recessivas,
exceto nas deficiências do inibidor de C1, que são
autossômicas dominantes, e da properdina, ligada ao X.
- Essas deficiências resultam em opsonização defeituosa, fagocitose e lise
de patógenos, e também prejudicam a remoção de imunocomplexos.
- As principais consequências associadas são infecções
recorrentes e surgimento de doenças autoimunes (ex: LES, glomerulonefrite)
- As principais consequências associadas são infecções
recorrentes e surgimento de doenças autoimunes (ex: LES, glomerulonefrite)
- Essas deficiências resultam em opsonização defeituosa, fagocitose e lise
de patógenos, e também prejudicam a remoção de imunocomplexos.
- As deficiências hereditárias são autossômicas recessivas,
exceto nas deficiências do inibidor de C1, que são
autossômicas dominantes, e da properdina, ligada ao X.
- O sistema complemento é um sistema de proteínas que
possui 3 vias de ativação: clássica, alternativa e das lectinas.
- São distúrbios isolados de componentes ou os inibidores do
complemento, podendo ser hereditários ou adquiridos
- ANGIOEDEMA HEREDITÁRIO
- Causado por deficiência ou disfunção do inibidor de C1,
proteína que regula a via de ativação clássica do
complemento.
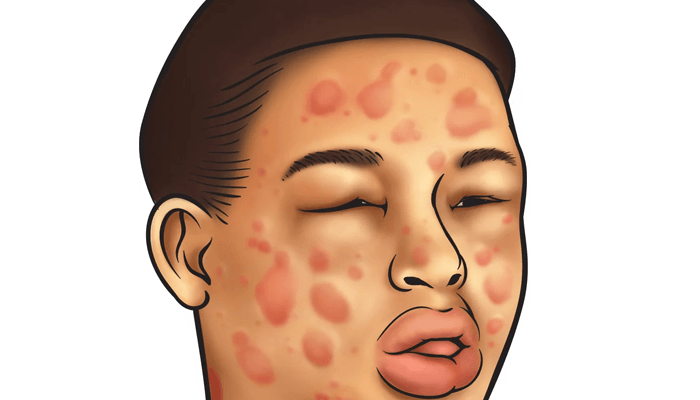
- Os sinais e sintomas do angioedema hereditário são
semelhantes aos das outras formas de angioedema
mediado pela bradicinina, com edema assimétrico e
levemente doloroso que muitas vezes está presente na
face, lábios e/ou na língua.
- Também podem ocorrer manifestações que sugerem obstrução
intestinal, náuseas, vômitos e desconforto com cólicas
- Também podem ocorrer manifestações que sugerem obstrução
intestinal, náuseas, vômitos e desconforto com cólicas
- Os sinais e sintomas do angioedema hereditário são
semelhantes aos das outras formas de angioedema
mediado pela bradicinina, com edema assimétrico e
levemente doloroso que muitas vezes está presente na
face, lábios e/ou na língua.
- Causado por deficiência ou disfunção do inibidor de C1,
proteína que regula a via de ativação clássica do
complemento.
- As deficiências do sistema complemento são as mais raras,
englobando cerca de 2% dos casos.
- Imunodeficiência Humoral
- As imunodeficiências primárias (ou congênitas) podem ser divididas
em 4 tipos principais, de acordo com o componente imunológico
deficiente:
- Manifestam-se precocemente em bebês e crianças, surgindo
desde o seu nascimento. Podem ser assintomáticas ou
altamente letais no primeiro ano de vida.
Medienanhänge
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Möchten Sie kostenlos Ihre eigenen Mindmaps mit GoConqr erstellen? Mehr erfahren.