34250723
COAGULOPATIAS
- Hemostasia
- É um processo fisiológico que
tem como objetivo manter o
sangue em estado fluido dentro
dos vasos sanguíneos, sem que
haja hemorragia ou trombose
- Cascata de coagulação
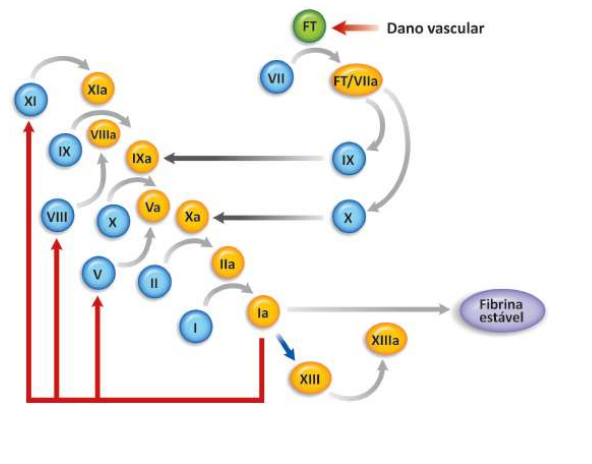
- Quando ocorre dano vascular, o fator tissular
(proteína transmembrana presente nas células do
subendotélio) é exposto, liga-se ao fator VII e
VIIa, iniciando o processo de coagulação.
- O complexo fator tissular (FT) - F VIIa - e Xa engatilha uma série de reações
bioquímicas de ativação e inativação da qual participam proteínas plasmáticas
(zimogênios de serinoproteases e cofatores), células (plaquetas e células
endoteliais) e íons (principalmente o cálcio). Este processo resulta na formação
de coágulo constituído por plaquetas e fibrina.
- O complexo fator tissular (FT) - F VIIa - e Xa engatilha uma série de reações
bioquímicas de ativação e inativação da qual participam proteínas plasmáticas
(zimogênios de serinoproteases e cofatores), células (plaquetas e células
endoteliais) e íons (principalmente o cálcio). Este processo resulta na formação
de coágulo constituído por plaquetas e fibrina.
- Quando ocorre dano vascular, o fator tissular
(proteína transmembrana presente nas células do
subendotélio) é exposto, liga-se ao fator VII e
VIIa, iniciando o processo de coagulação.
- Primaria
- Consiste na formação do tampão hemostático
pela agregação plaquetária. A lesão endotelial
expõe a matriz extracelular (MEC), favorecendo a
aderência e ativação plaquetária.
- A remoção do endotélio, por qualquer mecanismo, expõe o sangue ao contato com o colágeno da região
subendotelial, o que por si só promove a adesão das plaquetas na presença do fator vonWillebrand (fator
de coagulação VIII). Quando isto ocorre, as plaquetas tornam-se ativadas e liberam o conteúdo dos
grânulos citoplasmáticos. Entre outras substâncias, esses grânulos contêm adenosina-difosfato (ADP),
responsável pela ativação de outras plaquetas e pela modificação da sua forma. Além de ADP, esses
grânulos contém serotonina e tromboxane A2 (TXA2). Estas plaquetas ativadas vão se agregar umas às
outras formando um tampão que fornecerá a superfície adequada ao processo de coagulação do sangue,
produzindo um coágulo resistente.
- A remoção do endotélio, por qualquer mecanismo, expõe o sangue ao contato com o colágeno da região
subendotelial, o que por si só promove a adesão das plaquetas na presença do fator vonWillebrand (fator
de coagulação VIII). Quando isto ocorre, as plaquetas tornam-se ativadas e liberam o conteúdo dos
grânulos citoplasmáticos. Entre outras substâncias, esses grânulos contêm adenosina-difosfato (ADP),
responsável pela ativação de outras plaquetas e pela modificação da sua forma. Além de ADP, esses
grânulos contém serotonina e tromboxane A2 (TXA2). Estas plaquetas ativadas vão se agregar umas às
outras formando um tampão que fornecerá a superfície adequada ao processo de coagulação do sangue,
produzindo um coágulo resistente.
- Consiste na formação do tampão hemostático
pela agregação plaquetária. A lesão endotelial
expõe a matriz extracelular (MEC), favorecendo a
aderência e ativação plaquetária.
- Secundaria
- A coagulação sanguínea consiste na
conversão de uma proteína solúvel do
plasma, o fibrinogênio, em um polímero
insolúvel, a fibrina, por ação de uma
enzima denominada trombina.
- Intrinseca
- Na qual todos os
componentes estão
presentes no sangue.
- É desencadeada quando o fator XII e ativado pelo contato com alguma superfície carregada
negativamente (por exemplo, colágeno ou endotoxina). Além do fator XII, estão envolvidos
neste processo o fator XI, a pré-calicreína e o cininogênio de alto peso molecular (HMWK =
high molecular weight kinogen). Tanto o fator XI quanto a pré-calicreína necessitam da
HMWK para efetuar a adsorção à superfície em que está ligado o fator XIIa. Da interação
destes elementos é ativado o fator XI, que transforma o fator IX em IXa. O fator IXa e o fator
VIIa associam-se à superfície de fosfolipídio através de uma “ponte” de cálcio estimulando a
conversão de fator X para Xa.
- É desencadeada quando o fator XII e ativado pelo contato com alguma superfície carregada
negativamente (por exemplo, colágeno ou endotoxina). Além do fator XII, estão envolvidos
neste processo o fator XI, a pré-calicreína e o cininogênio de alto peso molecular (HMWK =
high molecular weight kinogen). Tanto o fator XI quanto a pré-calicreína necessitam da
HMWK para efetuar a adsorção à superfície em que está ligado o fator XIIa. Da interação
destes elementos é ativado o fator XI, que transforma o fator IX em IXa. O fator IXa e o fator
VIIa associam-se à superfície de fosfolipídio através de uma “ponte” de cálcio estimulando a
conversão de fator X para Xa.
- Na qual todos os
componentes estão
presentes no sangue.
- Extrinseca
- Na qual é necessária a presença da
proteína da membrana celular
subendotelial, o fator tecidual.
- A coagulação é desencadeada quando os
tecidos lesados liberam o fator tecidual
(tromboplastina tecidual), que forma um
complexo com o fator VII, mediado por íons
cálcio. Este complexo age sobre o fator X
estimulando sua conversão em Xa.
- A coagulação é desencadeada quando os
tecidos lesados liberam o fator tecidual
(tromboplastina tecidual), que forma um
complexo com o fator VII, mediado por íons
cálcio. Este complexo age sobre o fator X
estimulando sua conversão em Xa.
- Na qual é necessária a presença da
proteína da membrana celular
subendotelial, o fator tecidual.
- Comum
- Nesse ponto as duas vias encontram um caminho comum
em que ocorre a conversão de protrombina em trombina
que, por sua vez, estimula a transformação de fibrinogênio
em fibrina. A trombina cliva o fibrinogênio circulante em
fibrina insolúvel, formando uma rede de fibrina e induzindo
o recrutamento e ativação de mais plaquetas, formando um
tampão permanente, um trombo sólido.
- Nesse ponto as duas vias encontram um caminho comum
em que ocorre a conversão de protrombina em trombina
que, por sua vez, estimula a transformação de fibrinogênio
em fibrina. A trombina cliva o fibrinogênio circulante em
fibrina insolúvel, formando uma rede de fibrina e induzindo
o recrutamento e ativação de mais plaquetas, formando um
tampão permanente, um trombo sólido.
- Intrinseca
- A coagulação sanguínea consiste na
conversão de uma proteína solúvel do
plasma, o fibrinogênio, em um polímero
insolúvel, a fibrina, por ação de uma
enzima denominada trombina.
- É um processo fisiológico que
tem como objetivo manter o
sangue em estado fluido dentro
dos vasos sanguíneos, sem que
haja hemorragia ou trombose
- Coagulopatias hereditárias
- Hemofilias
- Etiologia
- São doenças hemorrágicas resultantes da deficiência do fator VIII (hemofilia A) ou IX (hemofilia B) da coagulação,
doenças genéticas de herança recessiva ligada ao cromossomo X, decorrente de mutações nos genes que codificam
os fatores VIII e IX da coagulação.
- Sua transmissão ocorre quase que exclusivamente a indivíduos do sexo masculino por mãe
portadora (cerca de 70% dos casos). Porém, em cerca de 30% dos casos, a doença origina-se a
partir de mutação nova, evento que pode acometer a mãe ou o feto.
- São doenças hemorrágicas resultantes da deficiência do fator VIII (hemofilia A) ou IX (hemofilia B) da coagulação,
doenças genéticas de herança recessiva ligada ao cromossomo X, decorrente de mutações nos genes que codificam
os fatores VIII e IX da coagulação.
- Clasificação
- O nível normal de atividade
coagulante dos fatores VIII e IX é
definido como 100%
- Grave
- <1% da normalidade
- <1% da normalidade
- Moderada
- 1%-5% da normalidade
- 1%-5% da normalidade
- Leve
- >5%-<40% da normalidade.
- >5%-<40% da normalidade.
- O nível normal de atividade
coagulante dos fatores VIII e IX é
definido como 100%
- Manifestações clinicas
- Mais comuns
- hemartroses
- manifestações hemorrágicas mais características das
formas graves da doença, atingindo mais frequentemente
as articulações do joelho, tornozelo, cotovelo, ombro e
coxo-femoral
- manifestações hemorrágicas mais características das
formas graves da doença, atingindo mais frequentemente
as articulações do joelho, tornozelo, cotovelo, ombro e
coxo-femoral
- hematomas
- hemartroses
- hematúria
- epistaxe
- melena/hematêmese
- sangramentos retroperitoniais
- intracraniano
- Mais comuns
- Diagnóstico
- Suspeita
- TTPA prolongado
- TTPA prolongado
- Fecha o diagnóstico
- dosagem dos fatores VIII e IX no plasma.
- Entretanto, o diagnóstico correto da condição de portadora de hemofilia (presente em cerca de 70%
das mães de pacientes) é mais difícil, uma vez que envolve o emprego de técnicas de biologia
molecular, metodologia esta mais complexa e de alto custo.
- dosagem dos fatores VIII e IX no plasma.
- Suspeita
- Tratamento
- Infusão venosa de concentrados de fator VIII ou IX de origem plasmática (Tabela 1) ou recombinante
por tempo variável, conforme gravidade e local da hemorragia.
- Do tipo A em casos leves
- Podem responder à infusão de desmopressina (0,3
microgramas/kg, diluído em 50 mL de solução salina,
endovenoso por 20 minutos) a cada 24 horas por, no máximo,
3-5 dias.
- Esse composto é análogo do hormônio antidiurético, aumenta a liberação
de fator VIII/FVW dos seus reservatórios.
- Esse composto é análogo do hormônio antidiurético, aumenta a liberação
de fator VIII/FVW dos seus reservatórios.
- Podem responder à infusão de desmopressina (0,3
microgramas/kg, diluído em 50 mL de solução salina,
endovenoso por 20 minutos) a cada 24 horas por, no máximo,
3-5 dias.
- O uso de antifibrinolíticos, tal como o ácido tranexâmico (25 mg/kg, via oral, a cada oito horas) ou ácido épsilon aminocaproico (EACA, 200 mg/kg,
via oral a cada seis horas) por 3-7 dias, é recomendado como adjuvante no tratamento de sangramentos principalmente mucosos, exceto nos
casos de hematúria
- A adoção de medidas locais, como compressas de gelo e pressão no local do sangramento, assim como
repouso e fisioterapia, é também recomendada conforme o caso.
- Infusão venosa de concentrados de fator VIII ou IX de origem plasmática (Tabela 1) ou recombinante
por tempo variável, conforme gravidade e local da hemorragia.
- Etiologia
- Doença de von Willebrand
- Etiologia
- Doença hemorrágica resultante de defeito quantitativo e/ou qualitativo do FVW
- Herdada como caráter autossômico dominante, resultante de mutações no gene que
codifica o FVW, localizado no cromossomo 12
- Doença hemorrágica resultante de defeito quantitativo e/ou qualitativo do FVW
- Sintomas
- frequentemente sangramentos cutâneo-mucosos
- equimoses
- epistaxe
- menorragia
- hemorragia gengival e digestiva
- Em casos mais graves
- hemartroses
- hematomas e hemorragia intracraniana
- hemartroses
- frequentemente sangramentos cutâneo-mucosos
- Diagnóstico
- Pode ser difícil
- Dosagem do cofator ristocetina (talves haja necessidade de repetição)
- Dosagem do FVW
- Dosagem do cofator ristocetina (talves haja necessidade de repetição)
- Obs: o TPPA normal não
exclui o diagnóstico da
doença
- Pode ser difícil
- Tratamento
- Objetivo
- Correção do FVW para além de 50% de
atividade coagulante na vigência de
hemorragia
- Correção do FVW para além de 50% de
atividade coagulante na vigência de
hemorragia
- desmopressina
- Para pacientes responsivos e com hemorragias de leve
a moderada intensidade.
- Para pacientes responsivos e com hemorragias de leve
a moderada intensidade.
- Infusão de concentrado de fator VIII contendo FVW
- Pacientes com DVW tipos 1 e 2A não
responsivos à desmopressina, para aqueles que
apresentam tipos 2B, 2N e 3 ou qualquer
subtipo mediante hemorragias graves
- Pacientes com DVW tipos 1 e 2A não
responsivos à desmopressina, para aqueles que
apresentam tipos 2B, 2N e 3 ou qualquer
subtipo mediante hemorragias graves
- Objetivo
- Etiologia
- Hemofilias
- Defeitos plaquetários
- Púrpura trombocitopênica idiopática
- Fisiopatologia
- Doença hemorrágica autoimune, caracterizada por anticorpos contra as
plaquetas do próprio paciente, que são, então, destruídas por fagocitose
principalmente no baço.
- Em crianças, a doença é aguda e, em geral, acompanha infecção viral.
- Doença hemorrágica autoimune, caracterizada por anticorpos contra as
plaquetas do próprio paciente, que são, então, destruídas por fagocitose
principalmente no baço.
- Clinica (sinais e sintomas)
- Epistaxe
- Petéquias
- Púrpuras e equimoses no corpo
- Menorragia em mulheres.
- Epistaxe
- Diagnóstico
- Exclusão de outras causas de plaquetopenia
- Hemograma
- Plaquetopenia
- Casos graves
- Contagem de plaquetas menor que 15.000/mm3
- Contagem de plaquetas menor que 15.000/mm3
- Casos graves
- Plaquetopenia
- Mielograma
- em pacientes > 60 anos
- Para diagnóstico diferencial com mielodisplasia e outras doenças
- Para diagnóstico diferencial com mielodisplasia e outras doenças
- em pacientes > 60 anos
- Sorologia para HCV
- Sorologia para HIV 1 e 2
- Diagnóstico diferencial
- Diagnóstico diferencial
- Exclusão de outras causas de plaquetopenia
- Tratamento
- Pacientes assintomáticos com plaquetas entre 30.000-40.000/mm3
- Acompanhamento clinico com contagem periódica de plaquetas (semanal ou quinzenal)
- Acompanhamento clinico com contagem periódica de plaquetas (semanal ou quinzenal)
- Paciente com contagem plaquetária
menor que 20.000/mm3
- Iniciar prednisona, 1 mg/kg/dia por 3-4 semanas até resposta ou aparecimento de efeitos colaterais, devendo a dose ser reduzida lentamente até sua retirada. Cerca
de 80% dos pacientes apresentam resposta após duas a três semanas de tratamento, embora a maioria dos pacientes apresentem recidiva com redução da dose.
- Iniciar prednisona, 1 mg/kg/dia por 3-4 semanas até resposta ou aparecimento de efeitos colaterais, devendo a dose ser reduzida lentamente até sua retirada. Cerca
de 80% dos pacientes apresentam resposta após duas a três semanas de tratamento, embora a maioria dos pacientes apresentem recidiva com redução da dose.
- Em casos de sangramento ativo ou pré-operatório
- Imunoglobulina venosa IGV, 1 g/kg/dia durante 2-3 dias
- Imunoglobulina venosa IGV, 1 g/kg/dia durante 2-3 dias
- Pacientes assintomáticos com plaquetas entre 30.000-40.000/mm3
- Fisiopatologia
- Púrpura trombocitopênica trombótica
- Púrpura trombocitopênica idiopática
- Doenças hemorrágicas devido a doenças sistêmicas.
- Insuficiência hepatica
- Fisiologia
- O fígado é fundamental para a hemostasia, sendo o principal
órgão de síntese dos fatores da coagulação (todos, exceto o
FVW), das proteínas anticoagulantes naturais (proteínas C e S) e
de proteínas relacionadas à fibrinólise.
- O fígado é fundamental para a hemostasia, sendo o principal
órgão de síntese dos fatores da coagulação (todos, exceto o
FVW), das proteínas anticoagulantes naturais (proteínas C e S) e
de proteínas relacionadas à fibrinólise.
- Diagnóstico
- TP e TTP encontram-se prolongados na
doença hepática avançada
- As plaquetas em número normal ou reduzido
- O TP é o teste mais sensível nas
fases iniciais da doença.
- TP e TTP encontram-se prolongados na
doença hepática avançada
- Tratamento
- Sangramento ativo
- Infusão de plasma fresco congelado (1015 ml/kg até de 8/8 horas)
- Infusão de plasma fresco congelado (1015 ml/kg até de 8/8 horas)
- Quando há plaquetopenia
- A transfusão de plaquetas (1 unidade para cada 10 kg)
- A transfusão de plaquetas (1 unidade para cada 10 kg)
- Nos casos de colestase
- Administração parenteral de vitamina K (10 mg, endovenosa)
- Administração parenteral de vitamina K (10 mg, endovenosa)
- Sangramento ativo
- Fisiologia
- Uremia
- Deficiência de vitamina K
- Septicemia
- Insuficiência hepatica
- BIBLIOGRAFIA: • Rezende, S. M. (2010). Distúrbios da hemostasia: doenças hemorrágicas. Rev Med Minas Gerais,
20(4), 534-53. • DE AZEVEDO, Maria Regina Andrade. Hematologia Básica: fisiopatologia e diagnóstico
laboratorial. Thieme Revinter Publicações LTDA, 2019. • LORENZI, Therezinha F. et al. Manual de hematologia
propedêutica e clínica. In: MANUAL DE HEMATOLOGIA PROPEDEUTICA E CLINICA. 1992. p. 500-500. • FAILACE,
Renato. Hemograma: manual de interpretação. Artmed Editora, 2015.
Medienanhänge
{kind=link}
Möchten Sie kostenlos Ihre eigenen Mindmaps mit GoConqr erstellen? Mehr erfahren.