346648
Coagulopatías hereditarias
- HEMOFILIA
- Enfermedad ligada al sexoDeficiencia
cuantitava ó funcional de algunos de los
factores de la cascada de coagulacion
- Hem A; def. factor VIII.
80% de los casos
- Hem B; def. factor IX
- Alt. en uno de estos
factores impide
formacion complejo
TENASA em la
superficie plaquetaria.
- DISM: TROMBINA
- NO HAY FORMACION DE COAGULO.
- NO HAY FORMACION DE COAGULO.
- DISM: TROMBINA
- Alt. en uno de estos
factores impide
formacion complejo
TENASA em la
superficie plaquetaria.
- Hem C; def factor XI
- Hem A; def. factor VIII.
80% de los casos
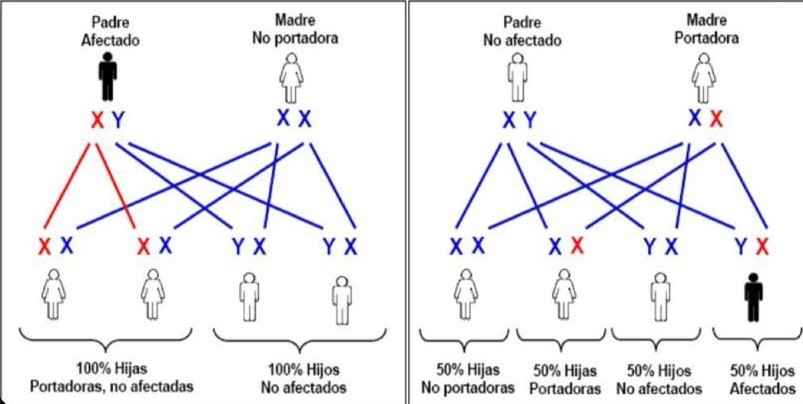
- Hemofilia A y B; ligadas al cromosoma X.
Hemofilia C; autosomica recesiva se
prsenta en hombres y mujeres.
- CLASIFICACION HEM A
Y B: leve (5-40%),
moderada (1-5%), grave
(menos de 1%)
- DIAGNOSTISCO DE HEM A y
B en paciente SINTOMATICO;
TTPA prolongada, TP y
plaquetas;normal. realizar
recuento de VIII,IX,FvW. (para
confirmacion)
- 1° TTO: Liofilizados de plasma de
FVIII,FvW debe acompañar para
que no disminuya el FVIII
- Pueden existir
inhibidores para el
factor administrado,
los que corresponden
a aloanticuerpos IgG
- Pueden existir
inhibidores para el
factor administrado,
los que corresponden
a aloanticuerpos IgG
- 2°TTO: Criprecipitado de plasma
resco congelado, 80-100 U F VIII y
FvW, fibrinogeno y FXIII
- 3°TTO: Plasma fresco congelado;
todos los factores de la coagulacion.
- Terapia alternativa; Desmopresina,
analogo de vasopresina que
aumenta FVIII,FvW.
- Pueden existir
inhibidores para el
factor administrado, los
que corresponden a
aloanticuerpos IgG
- Pueden existir
inhibidores para el
factor administrado, los
que corresponden a
aloanticuerpos IgG
- 1° TTO: Liofilizados de plasma de
FVIII,FvW debe acompañar para
que no disminuya el FVIII
- SIGNOS CLINICOS: Hemartrosis, hemorragias graves,
hematomas musculares, equimosis,hematuria.
- CLASIFICACION HEM A
Y B: leve (5-40%),
moderada (1-5%), grave
(menos de 1%)
- Pueden existir mujeres hemofilicas sí el
padre es hemofilico y la madre portadora,
pero esto es de escasa frecuencia. CASOS
ESPECIALES; mujeres con factor VIII o IX
niveles <50%, deben ser tratadas como
hemofilicas pero no lo son.
- EMBARAZADAS: fisiologicamnete
aumentan FVIII y FvW, se realiza de
rutina el TTPA, VIII, IX,FvW.
- EMBARAZADAS: fisiologicamnete
aumentan FVIII y FvW, se realiza de
rutina el TTPA, VIII, IX,FvW.
- Enfermedad ligada al sexoDeficiencia
cuantitava ó funcional de algunos de los
factores de la cascada de coagulacion
- EVW
- FvW :Secretado por cel. endoteliales, se
almacena en granulos alfa de plaquetas y
cuerpos de weibel-palade.Potenciador de
FVIII, que mantiene cu conecntraciion viable
en el plasma.
- FvW: Molécula multímera con sitios
de union para PLAQUETAS
(GpIb,GpIIb/IIIa), FVIII,colágeno.
- Trastorno cuantitativo o cualitativo
del FvW trastorno autosomico
recesivo ó dominante. cromosoma 12.
- Deficiencia cuantitativa parcial del
FvW (tipo1),autosomica dominante.
- disminución de los niveles plasmáticos del
FvW de entre un 15% a 50%, pero no existe
anormalidad en su función.
- disminución de los niveles plasmáticos del
FvW de entre un 15% a 50%, pero no existe
anormalidad en su función.
- deficiencia cualitativa (tipo 2) ,
Autosomica dominante.
- Subtipo 2A;deficiencia de los
multímeros de alto peso molecular
GPIb
- Subtipo 2B;
FvW--Plaquetas es
normal,
FvW---plasmatico no
presenta multimeros
lo que dism. afinidad
por GPIb
- Subtipo 2M; FvW---plaquetas
disminuida por mutacion del
dominio A1 lo que ocasiona
baja afinidad por GPIbalfa.,FvW
plasmatico presenta mulmeros
normales.
- Subtipo 2N;deficiente
formación del complejo
FvW-FVIII, por lo que los
pacientes que sufren
este trastorno presentan
un déficit de FVIII con
función plaquetaria
normal
- Subtipo 2A;deficiencia de los
multímeros de alto peso molecular
GPIb
- ausencia total del factor
(tipo 3) autosomica recesiva
- hay ausencia total del FvW en subendotelio,
plasma y plaquetas. Debido a esta
deficiencia, se ve afectado a su vez el
transporte y estabilidad del FVIII por lo que
los pacientes presentan alteración en la
hemostasia primaria y de la coagulación
sanguínea
- hay ausencia total del FvW en subendotelio,
plasma y plaquetas. Debido a esta
deficiencia, se ve afectado a su vez el
transporte y estabilidad del FVIII por lo que
los pacientes presentan alteración en la
hemostasia primaria y de la coagulación
sanguínea
- SIGNOS CLINICOS: debe de considerar la
historia clínica familiar •Lo más frecuente es
el angrado mucocutáneo; epistaxis,
gingivorragias, hematuria, hematemesis,
melenas. Puede haber menorragia •Períodos
sintomáticos y asintomáticos
- Deficiencia cuantitativa parcial del
FvW (tipo1),autosomica dominante.
- DE RUTINA; Tiempo sangria de Ivy
PROLONGADO,TTPA,PT. ESPECIFICAS;Factor de von
Willebrand antigénico (FvW:Ag) -Actividad del FvW
como cofactor de la Rsitocetina (FvW:CoR) -Actividad
coagulante del FVIII (FVIII:C) - agregacion plaquetaria
inducida con ristocetina
- TTO: ac.Tranexamico,
anticonceptivos
orales,Desmopresina, Liofilizado de
factor VIII
- TTO: ac.Tranexamico,
anticonceptivos
orales,Desmopresina, Liofilizado de
factor VIII
- FvW :Secretado por cel. endoteliales, se
almacena en granulos alfa de plaquetas y
cuerpos de weibel-palade.Potenciador de
FVIII, que mantiene cu conecntraciion viable
en el plasma.
Medienanhänge
{kind=link}
Möchten Sie kostenlos Ihre eigenen Mindmaps mit GoConqr erstellen? Mehr erfahren.