34061873
Description
Mind Map by CARLA MARIA ARAGON CONTRERAS, updated more than 1 year ago
|
|
Created by CARLA MARIA ARAGON CONTRERAS
about 3 years ago
|
|
Enfermedades congénitas,
cáncer, enfermedades
metabólicas y mitocondriales
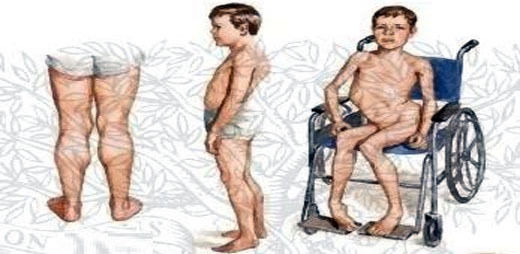
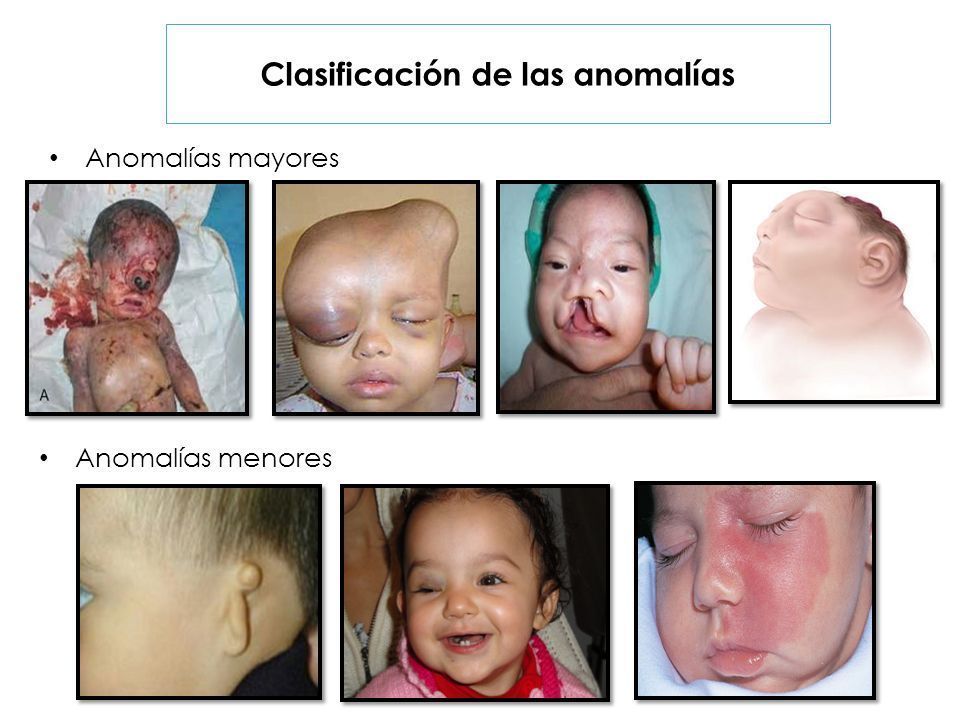
- Enfermedades congénitas
- Anormalidades de estructuras
anatómicas que son visibles en el
examen clínico y físico del neonato,
se hace evidente el defecto funcional
de un órgano afectado
anatómicamente.
- Pueden ocurrir debido a causas
genéticas o ambientales (pej. Zika).
Sin embargo, no todos los defectos
congénitos tienen una causa
genética, y no todas las
enfermedades genéticas presentan
defectos congénitos.
- Tipos
- Mayores
- Compromiso funcional importante para la vida del
paciente, hay consecuencias médicas, estéticas, se
requiere atención temprana en ocasiones de urgencia.
Tienen repercusión social (frecuencia 2-3% de RN)
- Compromiso funcional importante para la vida del
paciente, hay consecuencias médicas, estéticas, se
requiere atención temprana en ocasiones de urgencia.
Tienen repercusión social (frecuencia 2-3% de RN)
- Menores
- Defectos que denotan crecimiento desproporcionado a
una parte anatómica que no tienen generalmente un
significado relevante en la atención médica ni
significado social especial. Tiene una frecuencia en el
15% de neonatos.
- Defectos que denotan crecimiento desproporcionado a
una parte anatómica que no tienen generalmente un
significado relevante en la atención médica ni
significado social especial. Tiene una frecuencia en el
15% de neonatos.
- Mayores
- Diagnóstico
- Se basa principalmente en el interrogatorio, construcción del árbol
genealógico, examen físico del paciente y familiares, historia clínica
genética de la familia y paciente, exámenes complementarios o especiales
genéticos, revisión de literatura actualizada, consulta con especialistas y
medicina basada en evidencias.
- Se basa principalmente en el interrogatorio, construcción del árbol
genealógico, examen físico del paciente y familiares, historia clínica
genética de la familia y paciente, exámenes complementarios o especiales
genéticos, revisión de literatura actualizada, consulta con especialistas y
medicina basada en evidencias.
- Diagnóstico prenatal
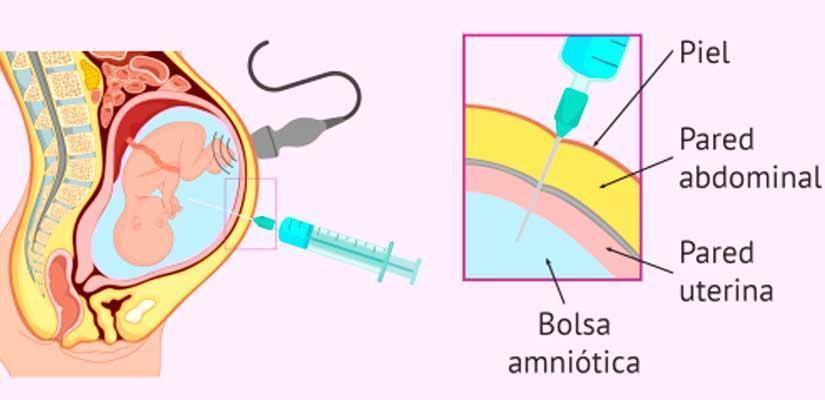
- Amniocentesis
- Por medio de una jeringa guíada por ultrasonido se
obtiene líquido amniótico vía transabdominal. Se
realiza de 14-16 semanas hasta las 20 semanas.
- El líquido posee células que contienen ADN fetal, pueden
determinarse enzimas y alfa-fetoproteína por medio de
PCR y otras. El riesgo de pérdida fetal es de 0.5% y
aumenta con la edad.
- El líquido posee células que contienen ADN fetal, pueden
determinarse enzimas y alfa-fetoproteína por medio de
PCR y otras. El riesgo de pérdida fetal es de 0.5% y
aumenta con la edad.
- Por medio de una jeringa guíada por ultrasonido se
obtiene líquido amniótico vía transabdominal. Se
realiza de 14-16 semanas hasta las 20 semanas.
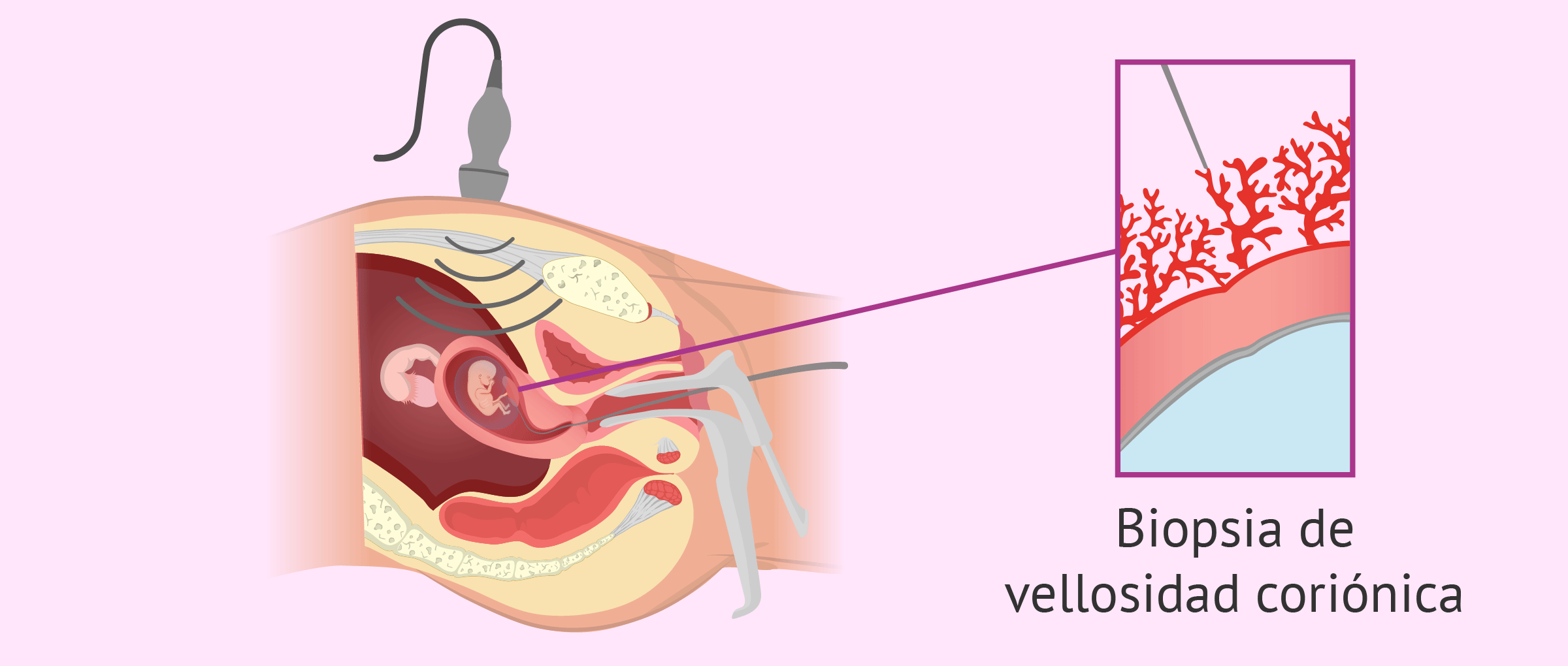
- Biopsia de vellosidades coriónicas
- Análisis de tejido placentario con tejido trofoblástico
fetal. Se realiza a las 9-12 semanas. Puede y debe
realizarse en etapas tempranas de gestación.
- Utiliza el mismo material para realizar los mismos
estudios de la amniocentesis, a excepción de la
alfa-fetoproteína. El riesgo de pérdida fetal es igual o
menos del 1%
- Utiliza el mismo material para realizar los mismos
estudios de la amniocentesis, a excepción de la
alfa-fetoproteína. El riesgo de pérdida fetal es igual o
menos del 1%
- Análisis de tejido placentario con tejido trofoblástico
fetal. Se realiza a las 9-12 semanas. Puede y debe
realizarse en etapas tempranas de gestación.
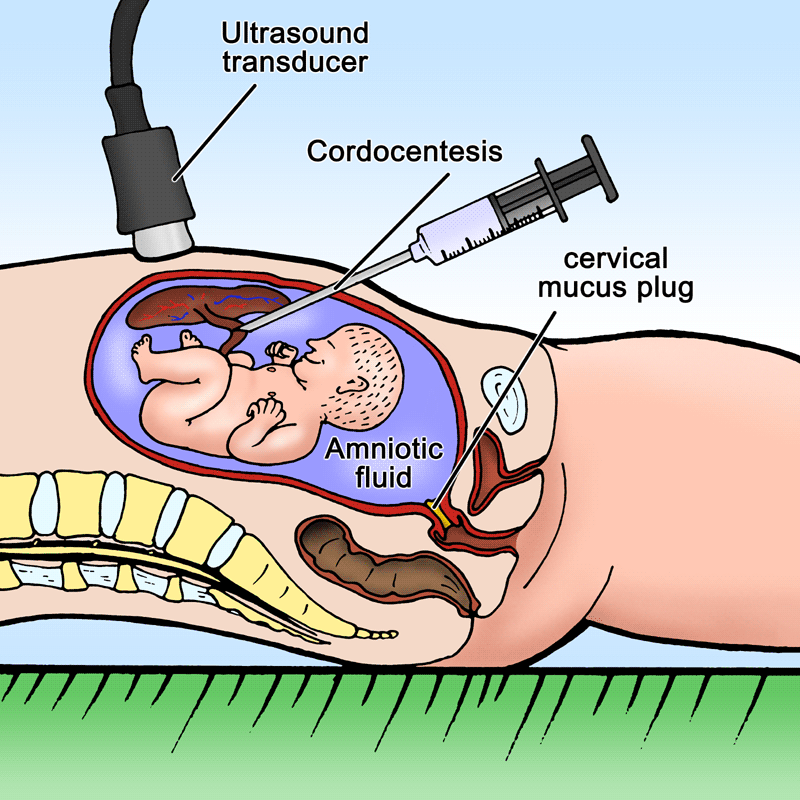
- Cordoncentesis
- Una aguja fina guiada por ultrasonido extrae sangre de la vena umbilical del feto. Se
realiza en gestaciones de más de 18 semanas. Se obtienen células para estudio de ADN
y proteínas cuando el cultivo de líquido amniótico falla, alteraciones solo identificadas
por bioquímica de plasma o células fetales y los resultados se precisan en menos de
una semana.
- El riesgo de pérdida fetal es de 1-2%.
- El riesgo de pérdida fetal es de 1-2%.
- Una aguja fina guiada por ultrasonido extrae sangre de la vena umbilical del feto. Se
realiza en gestaciones de más de 18 semanas. Se obtienen células para estudio de ADN
y proteínas cuando el cultivo de líquido amniótico falla, alteraciones solo identificadas
por bioquímica de plasma o células fetales y los resultados se precisan en menos de
una semana.
- Amniocentesis
- Enfermedades monogénicas detectables
en diagnóstico prenatal
- Autosómicas dominantes
- Acondroplasia, Sx de Marfán, neurofibromatosis, distrofia
miotónica, enfermedad de Huntington
- Acondroplasia, Sx de Marfán, neurofibromatosis, distrofia
miotónica, enfermedad de Huntington
- Autosómicas recesivas
- Anemia de células falciformes, fenilcetonuria, enfermedad de Gaucher,
fibrosis quística.
- Anemia de células falciformes, fenilcetonuria, enfermedad de Gaucher,
fibrosis quística.
- Ligadas al cromosoma X
- Hemofilia, sx de X frágil, distrofia muscular de Duchenne, deficiencia de
Omitin transcarbamilasa, adrenoleucodistrofia
- Hemofilia, sx de X frágil, distrofia muscular de Duchenne, deficiencia de
Omitin transcarbamilasa, adrenoleucodistrofia
- Autosómicas dominantes
- Anormalidades de estructuras
anatómicas que son visibles en el
examen clínico y físico del neonato,
se hace evidente el defecto funcional
de un órgano afectado
anatómicamente.
- Cáncer
- Su desarrollo se relaciona a mutágenos ambientales,
mutación somática y predisposición hereditaria combinados.
En los cánceres familiares debido a que se heredan las
mutaciones hay un cambio constitucional en todas las
células aumentando la posibilidad de más mutaciones
somáticas que conducen a tumores.
- El riesgo es bajo, el 5% de cx de mama, ovario a
intestinal se heredan por mutaciones en genes
autosómicos dominantes con penetración incompleta.
- Desarrollo del cáncer
- Se debe principalmente a los oncogenes (genes que pueden
transformar malignamente a las células normales) y los genes
supresores tumorales (inhiben la proliferación celular deteniendo
la división e iniciando apoptosis). Algunos defectos mutagénicos
son el cigarrillo y el virus de papiloma humano.
- Genes supresores de tumores
- Codifican proteínas que restringen
el crecimiento y transformación
maligna, pueden inicial apoptosis o
procesos de reparación de ADN. Su
pérdida, se asocia a la
tumorigénesis. Actúan de forma
recesiva, es decir requiere pérdida
de ambas copias para el desarrollo
de la neoplasia. Mutaciones que los
inactivan, se encuentran en cx
esporádico y hereditario.
- Al menos 2 mutaciones son
necesarias en el gen supresor
para eliminar su actividad y
promover el desarrollo del
cáncer
- Al menos 2 mutaciones son
necesarias en el gen supresor
para eliminar su actividad y
promover el desarrollo del
cáncer
- Codifican proteínas que restringen
el crecimiento y transformación
maligna, pueden inicial apoptosis o
procesos de reparación de ADN. Su
pérdida, se asocia a la
tumorigénesis. Actúan de forma
recesiva, es decir requiere pérdida
de ambas copias para el desarrollo
de la neoplasia. Mutaciones que los
inactivan, se encuentran en cx
esporádico y hereditario.
- Proto-oncogenes a oncogenes
- Genes con capacidad de corromper las
propias actividades celulares y conducir
a un estado maligno. Su función
normal incluye promover el crecimiento
y diferenciación celular. Cuando mutan
se vuelven oncogenes que codifican
proteínas perdiendo el control de
crecimiento, estos actúan de manera
dominante. Pueden causar inestabilidad
genética, impedir apoptosis y promover
la propagación (metástasis)
- Una sola mutación activa al
oncogén y se promueve el
desarrollo del cáncer.
- Una sola mutación activa al
oncogén y se promueve el
desarrollo del cáncer.
- Genes con capacidad de corromper las
propias actividades celulares y conducir
a un estado maligno. Su función
normal incluye promover el crecimiento
y diferenciación celular. Cuando mutan
se vuelven oncogenes que codifican
proteínas perdiendo el control de
crecimiento, estos actúan de manera
dominante. Pueden causar inestabilidad
genética, impedir apoptosis y promover
la propagación (metástasis)
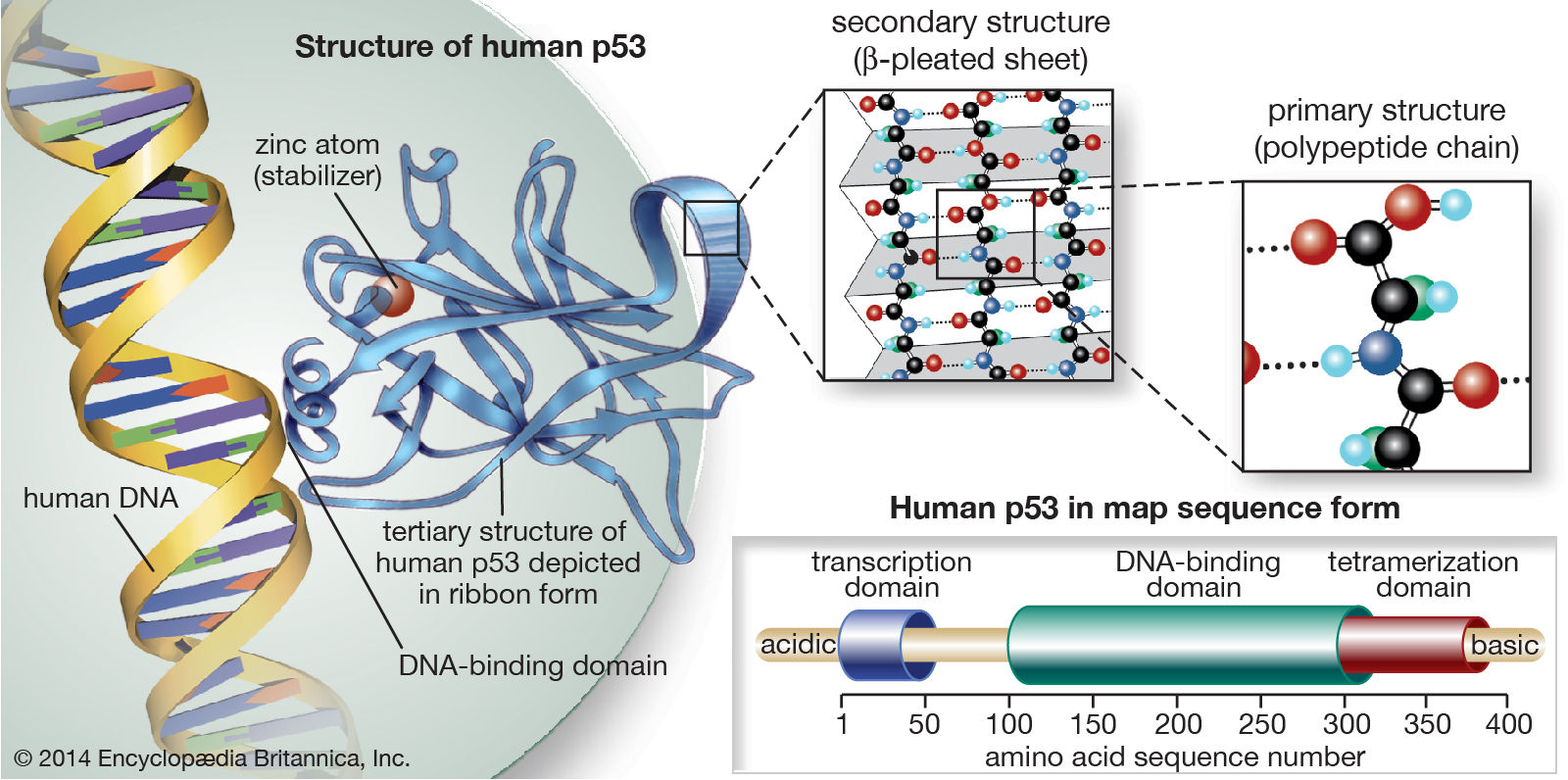
- Guardián del genoma
- TP53
- Gen supresor de tumores más asociado al
desarrollo del cáncer. Presenta
frecuentemente mutaciones en los humanos
(50% de todos los humanos tienen
mutaciones en el gen). Su falta induce un
trastorno llamado Li-Fraumeni (alta
incidencia cx mamario, cerebral y leucémico).
- Codifica la proteína p53 que regula el ciclo celular y la apoptosis.
- Codifica la proteína p53 que regula el ciclo celular y la apoptosis.
- Gen supresor de tumores más asociado al
desarrollo del cáncer. Presenta
frecuentemente mutaciones en los humanos
(50% de todos los humanos tienen
mutaciones en el gen). Su falta induce un
trastorno llamado Li-Fraumeni (alta
incidencia cx mamario, cerebral y leucémico).
- TP53
- Genes supresores de tumores
- Se debe principalmente a los oncogenes (genes que pueden
transformar malignamente a las células normales) y los genes
supresores tumorales (inhiben la proliferación celular deteniendo
la división e iniciando apoptosis). Algunos defectos mutagénicos
son el cigarrillo y el virus de papiloma humano.
- Desarrollo del cáncer
- El riesgo es bajo, el 5% de cx de mama, ovario a
intestinal se heredan por mutaciones en genes
autosómicos dominantes con penetración incompleta.
- Su desarrollo se relaciona a mutágenos ambientales,
mutación somática y predisposición hereditaria combinados.
En los cánceres familiares debido a que se heredan las
mutaciones hay un cambio constitucional en todas las
células aumentando la posibilidad de más mutaciones
somáticas que conducen a tumores.
- Errores innatos del
metabolismo o
enfermedades metabólicas
- Trastornos bioquímicos en la estructura o
función de proteínas originado por
mutaciones en el ADN. Frecuentemente
son de tipo monogénicas y de herencia
autosómica recesiva. Existen al menos
más de 700.
- Pocas veces se presenta en el RN o
durante la primera infancia (menos del
50%), se hace presente la mayoría de
veces en la adultez. Su diagnóstico
precoz es vital para el éxito del
tratamiento.
- Pocas veces se presenta en el RN o
durante la primera infancia (menos del
50%), se hace presente la mayoría de
veces en la adultez. Su diagnóstico
precoz es vital para el éxito del
tratamiento.
- Consecuencias
- En ocasiones cuando no es útil el análisis de ARN o ADN o no es factible, se
detecta bioquímicamente por medio de la deficiencia de la enzima o
trastornos causados por proteína estructural defectuosa. Los errores
innatos del metabolismo son causa de muertes prematuras, trastornos
neurológicos como retraso mental, mala calidad de vida del paciente y sus
familaires.
- En ocasiones cuando no es útil el análisis de ARN o ADN o no es factible, se
detecta bioquímicamente por medio de la deficiencia de la enzima o
trastornos causados por proteína estructural defectuosa. Los errores
innatos del metabolismo son causa de muertes prematuras, trastornos
neurológicos como retraso mental, mala calidad de vida del paciente y sus
familaires.
- Diagnóstico
- Se precisan pruebas sensibles y específicas.
- Deben ser seguras, simples y económicas. Además, se requieren
programas para identificar parejas portadoras, diagnóstico
prenatal en el primer trimestre.
- Deben ser seguras, simples y económicas. Además, se requieren
programas para identificar parejas portadoras, diagnóstico
prenatal en el primer trimestre.
- Se precisan pruebas sensibles y específicas.
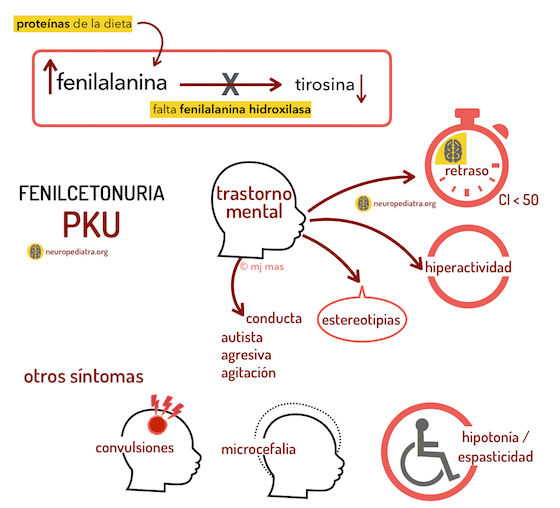
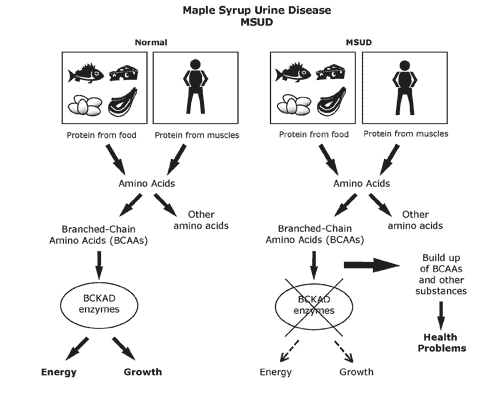
- Ejemplos
- Síndrome de Fnaconi, fibrosis quística del páncreas,
hemocromatosis, fenilcetonuria, tirosinemia, enfermedad
de la orina de jarabe de arce, galactosemia, acidemias
lácticas congénitas, tx de beta-oxidación, entre otros.
- Síndrome de Fnaconi, fibrosis quística del páncreas,
hemocromatosis, fenilcetonuria, tirosinemia, enfermedad
de la orina de jarabe de arce, galactosemia, acidemias
lácticas congénitas, tx de beta-oxidación, entre otros.
- Trastornos bioquímicos en la estructura o
función de proteínas originado por
mutaciones en el ADN. Frecuentemente
son de tipo monogénicas y de herencia
autosómica recesiva. Existen al menos
más de 700.
- Enfermedades mitocondriales
- Se transmite el ADNmt exclusivamente vía materna. Al
menos 1 en 5000 tienen una enfermedad mitocondrial,
algunos estudios demuestran que las mutaciones de 1
en 200 no implica una enfermedad. Así también, son
secundarias a defectos de la síntesis de ATP.
- 90% de la energía proviene de mitocondrias, al menos 3000 genes en
la mitocondria pueden encontrarse y 1500 en la fosforilación oxidativa.
El ADNmt tine 16.569pb en una cadena doble circular, cada célula
tiene cientos de mitocondrias y cada una tiene más de 10 copias de
ADNmt.
- Tipos de mutaciones mitocondriales
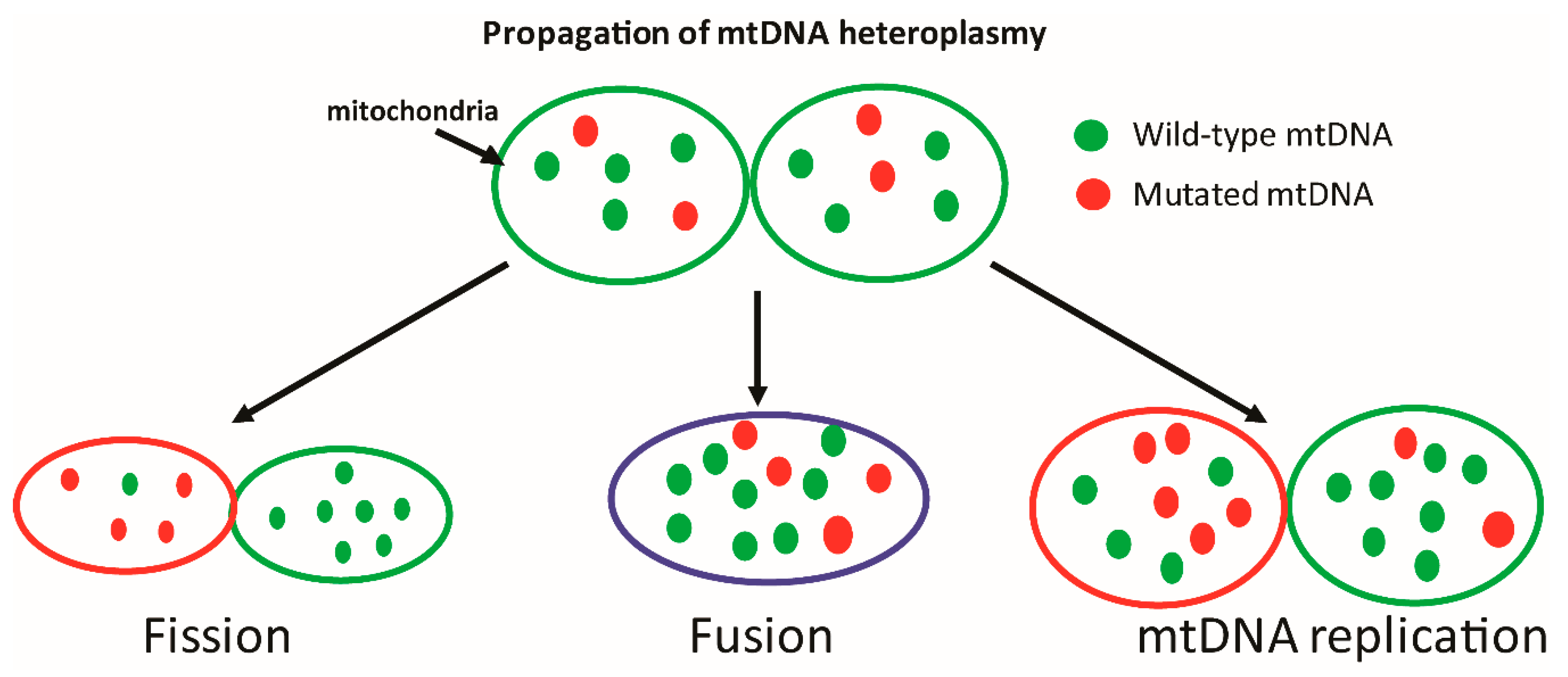
- Heteroplasmia
- Una sola célula tiene una mezcla
de ADNmt mutante y de tipo
salvaje.
- En más del 60% hay enfermedad
- En más del 60% hay enfermedad
- Una sola célula tiene una mezcla
de ADNmt mutante y de tipo
salvaje.
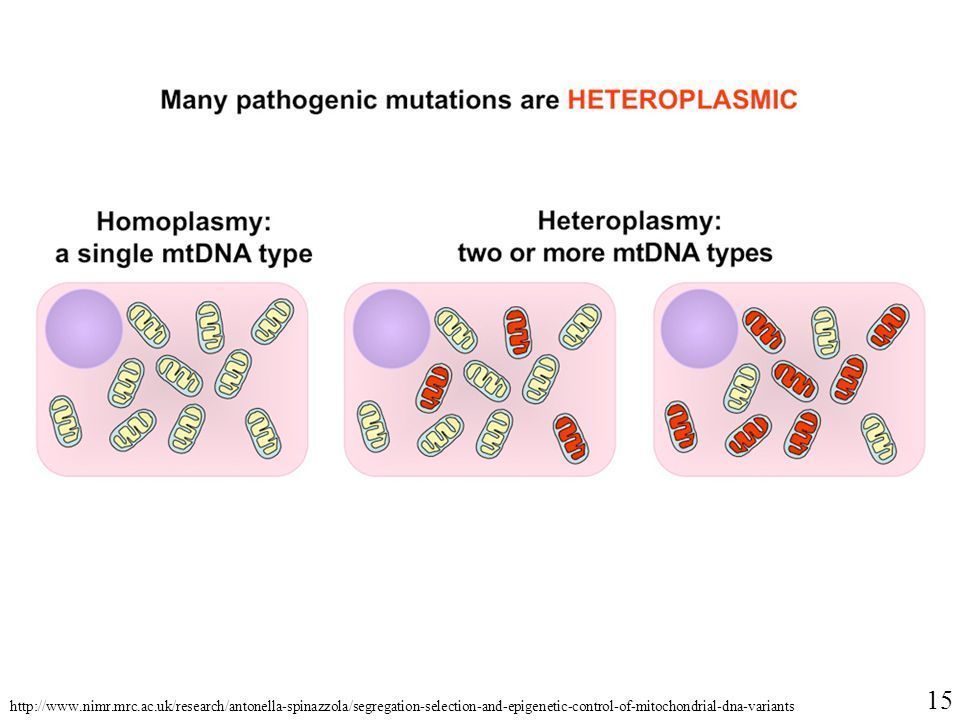
- Homoplasmia
- Dominio total de ADN mutante o normal.
- Dominio total de ADN mutante o normal.
- La gravedad depende de las
proporciones relativas de
ADN silvestre y mutante
presente.
- Muchas enfermedades se asocian a deleciones o mutaciones del
ADNmt. No posee intrones, la mayoría ocurre en forma esporádica y
las mutaciones son 5-10 veces más que en ADN nuclear, ya que no
se protege de histonas, tiene mecanismos de reparación deficientes,
expuesto a radicales libres. Pueden presentarse a cualquier edad, en
infantes son mortales.
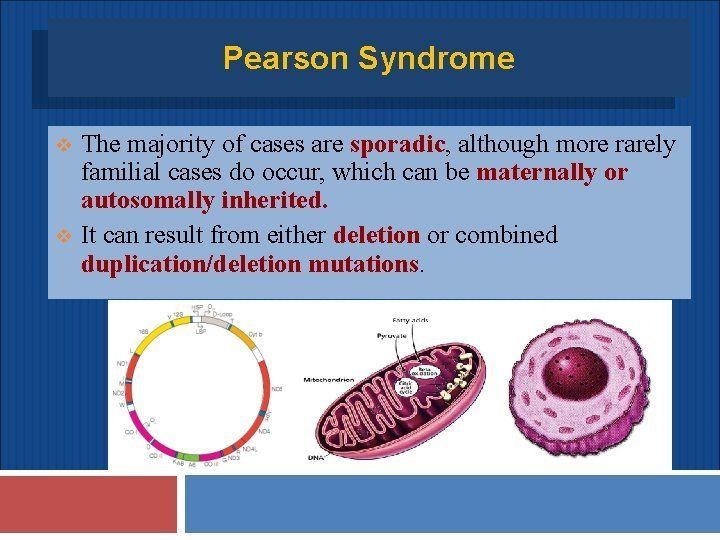
- Ejemplos
- Sx de Pearson, Sx de Leigh, Mitochondrial myopathy,
encephalopathy, lactic acidosis and stroke-like episodes
(MELAS), La neuropatía, ataxia y retinitis pigmentosa
(NARP), entre otras.
- Sx de Pearson, Sx de Leigh, Mitochondrial myopathy,
encephalopathy, lactic acidosis and stroke-like episodes
(MELAS), La neuropatía, ataxia y retinitis pigmentosa
(NARP), entre otras.
- Ejemplos
- Muchas enfermedades se asocian a deleciones o mutaciones del
ADNmt. No posee intrones, la mayoría ocurre en forma esporádica y
las mutaciones son 5-10 veces más que en ADN nuclear, ya que no
se protege de histonas, tiene mecanismos de reparación deficientes,
expuesto a radicales libres. Pueden presentarse a cualquier edad, en
infantes son mortales.
- Heteroplasmia
- Tipos de mutaciones mitocondriales
- 90% de la energía proviene de mitocondrias, al menos 3000 genes en
la mitocondria pueden encontrarse y 1500 en la fosforilación oxidativa.
El ADNmt tine 16.569pb en una cadena doble circular, cada célula
tiene cientos de mitocondrias y cada una tiene más de 10 copias de
ADNmt.
- Se transmite el ADNmt exclusivamente vía materna. Al
menos 1 en 5000 tienen una enfermedad mitocondrial,
algunos estudios demuestran que las mutaciones de 1
en 200 no implica una enfermedad. Así también, son
secundarias a defectos de la síntesis de ATP.
- Realizado por: Carla María
Aragón Contreras, 200-18-10367
Media attachments
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Want to create your own Mind Maps for free with GoConqr? Learn more.