3380605
| Frage | Antworten |
| Protein p53? | wenn die DNA reparatur nicht gelingt >ansteigende konz von phosphoryliertem p53 induziert apoptose. |
| Sorbitol, wie entsteht es? | ensteht durch reduktion von c1 der glucose und reduktion von c2 an fructose reduktion + 2h der carbonylgruppe, -> zuckeralkohol, bild=glycerol |
| aerobe glykolyse | in gegenwart von o2 gelangen pyruvat und NADH in MITOCHONDRIEN!! und da wird 2 pyruvat durch Pyruvatdehydrogenase weiter zu acetyl coa verstoffwechselt , während nadh durch die atmungskette reoxidiert wird ( für ATP gewinn) |
| anaerobe glykolyse: was wird gebildet? Corizyklus erklären bitte | einziger stoffwechselweg, der in abwesenheit von o2 ATP erzeugen kann, bei diesem weg müssen im ZYTOPLASMA aus pyruvat und nadh gärungsprodukte gebildet werden. (->> 2 lactat ), um NAD+ für den fortgang der glykolyse zu regenerieren. Leber kann das Lactat wieder aufnehmen und in Pyruvat umwandeln. Pyruvat wird dann über die Gluconeogenese (pyruvatcarboxylase, PEP-carboxykinase, fru-6-phosphatase-glu-6-phosphatase)in Glu umgewandelt. Für aufbau einer glu werden 2 lactat benötigt. so gelangt glu wieder in muskel-> lactat = corizyyklus |
| Glykolyse-Bilanz? | 2ATP 2 NADH 2Pyruvat |
| Glykolyse-Ablauf bis zu den 2 Triosen | Glu passiert durch GLUTS die zellmembran, im zytosol der zelle erfolgt die Hexokinase (bzw. insulinabh. Glikokinase in leber) phosphorylierung zu glu-6-ph, um sie in der zelle zu halten. IRREV - glu-6-phospha-isomerase wandelt glu-6-ph in fru-6-ph um -phosphofruktokinase phosphoriliert fru-6-phosphat zu fru-1,6- bisphosphat >ATP VERBRAUCH IRREV -Aldolase spaltet fru-1,6-bisphosphat in 2 triosen: dihydroxyacetonphosphat und glycerinaldehyd-3-phosphat also ein alkohol und ein aldehyd enstehen |
| Glykolyse-Ablauf ab den 2 Triosen | dihydroxyacetonphosphat und glycerinaldehyd-3-phosphat -Glyceral-3-pho-dehydrogenase oxidiert unterbildung von NADH glyceral-3-pho zu 1,3 bisphosphoglycerat, dabei wird pho. über säureanhydridbindung an c1 ins molekül aufgenommen>>substratkettenphosphorylierung -3PHOSPHOGLYCERAT-kinase überträgt ph auf ADP und spaltet somit die säureanhydridbindung, 3-phosphoglycerat und atp entstehen!!! -umlagerung zu 2-phosphoglycerat durch phosphoglycerat mutase -enolase spaltet wasser ab , sodass phosphoenolpyruvat entsteht (übertragungspotenzial der phosphatgruppe!) -pyruvatkinase spaltet das pho von phosphoenolpyruvat ab und überträgts auf ADP, wobei pyruvat entsteht und 2 ATP! IRREV!!!! |
| zu was wird glu in der glykolyse abgebaut? | Pyruvat o. Lactat |
| Substratkettenphosphorylierung, was für eine reaktion ist das? | eine energieliefernde reaktion, anorg. phosphat wird auf ein substrat übertragen (phosphorylierung), so kommts zur ausbildung von säuranhydridbindungen im substrat(sehr energiereich!) Nun kann diese energie wiederum genutzt werden um ADP zu phosphorylieren |
| Funktionsweise der atmungskette? | Aus den wasserstoffatomen der reduzierten coenzyme und dem sauerstoff den wir einatmen wird wasser gebildet, dabei wird energie frei die zur bildung von ATP genutzt wird. Energie wrd in einem protonengradienten gespeichtert, der über die innere mitomembran aufgebaut wird. die energie zum aufbau des gradienten stammt aus den reduzierten coenzymen von cz, beta ox, gylkolyse freigesetzt wird die energie dadurch, dass wasserstoffatome unter bildung von h2o auf sauerstoff übertragen werden. der protonengradient dient dazu , mttels ATP synthase ATP zu bilden. |
| inputs und outputs der atmungskette? | INPUTS NADH+H+ FADH2 O2 ADP Outputs: oxidierte coenzyme NAD+ und FAD, ATP, H20 |
| Was ist die Aufgabe der 4 Atemkettenkomplexe? | Die komplexe haben alle die aufgabe wasserstoffatome von den reduzierten coenzymen in intermembran raum zu pumpen-> protonenpumpe. die elektronen die durch die komplexe fließen, werden zum schluss auf o2 übertragen, gemeinsam mit den protonen aus den red. Coenzymen entsteht wasser> knallgasr. |
| Komplex 1 A.kette | Komplex 1: H+ atome werden von NADH+H+ auf ubichinon übertragen, komplex 1 enthält FMN und eisen schwefel komplexe als prostet. Gruppen komplex 1 besitzt von allen komplexen das neg. redoxpotenzial. |
| Komplex 2 der A.kette | Komplex2 H+ atome werden von succinat auf ubichinon übertragen, komplex 2 enthält FADH2 und eisen schwefel komplexe als prost. Gruppen, er ist keine protonenpumpe udn befindet sich an der innenseite der mitomembran (beim übertragen von h+ auch ubichinon wirds zum ubichinol reduziert. Ubichinol= sammelpooL für alle reduktionsäquivalente der atmungskette) //ubchinon kann 2 elektronen aufnehmen |
| Komplex 3 der A.kette | Komplex 3: elektronen werden auf cytochrom c übertragen , komplex 3 enthält cytochrom b und einsen schwefel komplexe als prost. Gruppen //cytrochrom kann ein elektron aufnehmen! |
| Komplex 4 der A.kette | elektronen werden von cytochrom c auf sauerstoff übertragen! cytochrom c wird zu O2- oxidiert, und diffundiert in die mito matrix, um sich dort mit 2 h+ zu h2O zu verbinden= knallgasreaktion!! |
| Was passiert mit den Elektronen, die durch die 4 Komplexe fließen?? | die elektronen die durch die komplexe geflossen sind, werden zum schluss auf o2 übertragen, gemeinsam mit den protonen aus den red. Coenzymen entsteht wasser> knallgasr. |
| ATP synthase A.kette, durch was wird sie angetrieben? Wie viel ATP entstehen? | atpsynthase: rückführung der in den intermembranraum gepumpten protone zurück in den matrixraum.=turbine, oxidative phosphorylierung von ADP= substratkettenph. BILANZ: pro oxidiertem NADH+H+ entstehn 2,5 ATP pro oxidiertem FADH2 entsten 1,5 ATP also insgesamt 4 ATP |
| Wodurch werden die Protonenpumpen der Komplexe angetrieben?? | Reduzierte Coenzyme werden oxidiert!Elektronenabgabe während den Komplexen! Die bei den oxidationen freigesetzte energie wird genutzt um protonen vom matrixraum in den intermemranraum zu pumpen (nur komplex 1,3,4) |
| Wie sieht das redoxpotenzial aus während der Atmungskette? | Redoxpotenzial wird während der atmungskette immer positiver! Elektronenabgabe der oxidierten Coenzyme. in den komplexen durchlaufen die H-atome/elektronen die spannungsreihe |
| ATP ausbeute bei volständigem GLU ABBAU? | 32ATP |
| Hemmstoffe der atmungskette: | barbiturate, komplex 1 wird gehemmt und produkthemmung |
| Knallgasreaktion-formel und energie? | ½ O2+H2-> H2o gibbs=-240kj/mol |
| Sinn der elektronentransportkette der Atmungskette? | die energie der knallgasr. wird nicht auf einmal freigesetzt-> sonst würde sie die zelle zerstören die energie wird in kleinen paketen freigesetzt, WIE? die elektronen, die bei dieser knallgasreaktion vom wasserstoff auf den sauerstoff übertragen werden , müssen vorher durch mehrere redoxsysteme fließen bevor sie den sauerstoff in komplex 4 erreichen! die atmungskette enthält 4 redoxsysteme, die alle unterschiedl. redoxpotenziale besitzen, also jeder komplex hat ne andere affinität zu elektronen. Die elektronen fließen vom komplex mit dem negativsten redoxpotenzial zu dem mit dem positivsten, die elektronen aus den reduzierten coenzymen werden auf diese weise von komplex 1 zu 4 transportiert. besonders wichtig ist dabei, dass bei den einzelnen elektronenübergängen frei werdende energie genutzt wird um protonen durch die membran zu pumpen! |
| was issn redoxsystem? | ein redoxsytsem ist ne verbindung die durch elektronenaufbnahme/abgabe, vom reduzierten in den oxidierten zustand übergehn kann und umgekehrt |
| A.kette: Woher kommt der 02? | Diesen Sauerstoff müssen wir ständig mit dem Blut hintransportieren. Dafür haben wir ja eine Lunge, rote Blutkörperchen und Hämoglobin.. der eingeatmete sauerstoff wird nicht zur bildung des ausgeatmeten co2 verwendet, sondern in den mitochondiren zur bildung von wasser-> atp synthese |
| Welche Farbe haben Mitochondrien? | Mitochondrien sind durch ihren gehalt an cytochrom stark braun gefärbt! |
| wie findet die oxidative phosphorylierung in braunem fettgewebe statt? | In braunem fettgewebe wird die oxidative phosphorylierung entkoppelt! brauen fettgwb> aufg= versorgung mit wärme, für diese wärmeversorgung gibt spez. Kanalproteine in der inneren mitomembran= thermogenin, welches für protonen durchlässig ist! Zusammenbruch des aufgebauten protonengradienten durch die atmungsk. Statt atp synthese wird wärme erzeugt! |
| Was passiert mit dem ganzen ATP was in der A.kette gemacht wird? | Als Energiequelle wird ATP für die grundlegenden energieverbrauchenden Prozesse aller Lebewesen genutzt: chemische Arbeit, wie Synthese organischer Moleküle, osmotische Arbeit, wie aktiver Stofftransport durch Biomembranen, sowie mechanische Arbeit, Bewegungen bei der Muskelkontraktion |
| Wie gelangt das ATP aus dem mitochondrium in das zytosol? | In der inneren mitomembran gibt es ATP/ADP translokatoren, atp wird im austausch von adp aus dem matrixraum befördert |
| Wie steckt in ATP so viel Energie? | Die Phosphate sind über Säureanhydrid-Bindungen miteinander verbunden. Werden diese Bindungen durch Enzyme hydrolytisch gespalten, entsteht das ADP und Orthophosphat und Pyrophosphat. Die Spaltung der Bindung verbraucht Energie; insgesamt werden jedoch durch die anschließende Hydratation des abgespalteten Phosphats unter Standardbedingungen jeweils 32,3 kJ/mol (Spaltung einer Bindung) Energie für Arbeitsleistungen in den Zellen frei. |
| Was ist das Redoxpotenial von 02? | 2V, hohe anziehungskaft auf elektronen! (Elektronenaufnahme = reduktion!) |
| Was ist der Unterschied zw NADH/H und FADH2? | FAD hat ein höheres redoxpotenzial als NAD d.h. höhere anziehungkraft auf elektronen |
| Wie viel ATP verstoffwechselt der Mensch? | jeder Mensch verstoffwechselt ungefähr so viel ATP pro Tag wie das eigene Körpergewicht |
| Endosymbiontentheorie?? | mitos sind wahrscheinl. Aus aeroben bakterien entstanden, die mit anaerob lebenden eukaryoten eine symbiose eingegangen sind, mitos besitzten eine ringförmige dna und eigene ribosomen!!dna kodiert für 13 proteine, 2 membrenen, innere membran vom ehemaligen symbionten-> prokaryot. Membran, enthält cardiolipin, innere mitomembran ist für O2 durchlssg |
| Was sind Kohlenhydrate ? Wie kommen sie zur stande? | Kohlenhydrate sind aldehyde oder ketone eines mehrwertigen alkohols mit der allg. formel Cn(H2O)n. sowohl ketone als auch aldehyde sind moleküle bei denen eine oh-gruppe oxidiert wurde. In dem fall der aldehyde wurde eine primäre hydroxylgruppe, bei den ketonen eine sekndäre hydroxylgruppe oxidiert. Bildung der beiden einfachen Zucker Dihydroxyaceton (Ketose) und Glycerinaldehyd (Aldose) aus dem Polyalkohol Glycerin (Glycerol) durch Abgabe von 2 Wasserstoffatomen. |
| Was ist der Hauotunterschied zw. verstoffwechselung von Glu und Fru? | Unterschied zu Glu, fru wird im gegensatz insulinunabhgg in die zelle aufgenommen-> glut5 transporter deshalb können diabetiker die an insulinmangel leiden, fru als glu ersatz zu sich nehmen. sowohl fru als auch sorbitol gelten als zuckerersatzstoffe, da sie insulinunabhgg verstoffwechslet werden, sorbit wird dabei vor der glykolyse in fru umgewandelt |
| wie verstoffwechselt ein Diabetiker Fruktose? | Fuctose aus der nahrung wird über nen besonderen weg (aldolase B)zu glyceral und glycerol 3 phosphat abgebaut. Reguliert durch ins und cAmp. Abbau von fru findet vor allem in der leber statt, mit dem ziel aus fru energie zu gewinnen. 1)in de leberzelle angekommen, wird fru durch durch fruktokinase zu fru-1-pho phosphoryliert. 2)durch aldolase B, zu glceron-3-pho und glyceral gespalten glyceron-3-pho kann unverändetr in die glykolyse eingeschleust werden. glyceral wird unter atpverbrauch phosphoryliert, als glyceral-3-ph kanns dann abgebaut werden. genau wie glykolyse liefert auch fru 2 atp in extrahepat.gwb wird nur ein kl. teil der fructose aufgenommen, wieder insulinunabhgg über glut5, der unterschied ist dass in diesen zellen nur hexokinase herrscht, sodass fructose zu fru-6-pho phosphroyliert wird und nicht zu fru-1-pho fru-6-pho wird auch in die glykolyse eingeschleust. |
| Fructosebildung? | Fructosebildung: Fructose kann über den Polyolweg aus glucose gebildet werden. Als zwischenprodukt tritt dabei sorbitol auf.-> aldehyd reduktase |
| Glykogen Struktur? | ist ein verzweigtes Homopolymer der glucose, die glucose reste sind alfa1->4 glykosidisch miteinander verknüpft, etwa jeder 10.rest ist über alfa1->6bindung mit einer weiteren glucose verbunden, das glykogen ist dann verzweigt |
| Glykogen dient wozu | dient als Kohlenhydratreserve, aus der bei bedarf glukosephosphate und glukose freigesetzt werden können. |
| Warum eignet sich Glykogen als Speicherform? Wo wirds gespeichert und wie viel? | Das unlösl. große Makromolekül glykogen ist osmotisch wenig aktiv und einigt sich deshlab als speicherform. es kann von allen seiten bearbeitet werden Der mensch kann max. 450g glykogen speichern, gespeichert wird in leber und muskeln. |
| Glykogen in der Leber, Fkt? | Das glykogen in der leber, dient der aufrechterhaltung des blutzuckerspiegels. Glykogengehalt der leber ist sehr variabel, bei hungerphasen fällt er auf fast null ab, danach übernimmt die gluconeogenese die versogung des organs mit glu. Das Leberglykogen wird nie vollst. Abgebaut, es werden immer nur die nicht reduzierenden enden verkürzt oder bei glykogensynthese verlängert |
| Glykogen in den Muskeln, Fkt? | Das glykogen in den muskeln dient diesen als energiereserve und ist nicht wie in der leber, bei der regulation des blutzuckerspiegels beteiligt. keine glucose-6-phosphatase im muskel!! Also muskeln können nicht glukose ins blut freisetzen! |
| Glykogensynthese in Muskeln und Leber | bei reichl. Gluangebot wird durch hexokinase glu zu glu-6-ph dann durch mutase zu glu-1-phosphat isomerisiert. 1)umandlung in UDP-glucose (Uridindiphosphat Glucose =aktivierte form, da das knüpfen glykosidischer bindungen endergon ist) und zur glykogensynthese verwendet 2)glykogen-synthase überträgt der reihe nach glucosereste von udp-glucose auf die nicht reduzierenden enden der vorhandenen zweige eines bestehenden glykogenmoleküls 3)ab über 11resten der kette, wird durch das verzewigende enzym ein oligosaccharid aus 6-7 resten an ihrm ende abgespalten und im inneren der kette in alfa1->6 bindung wieder hinzugefügt verzeigte struktur des glykogens ermöglicht die rasche freisetzung von glucoseresten |
| Glykogenabbau | Abbau von Glykogen zu Glukose-1-Phosphat und Glucose. Bei glukosemangel wird gespeichertes glykogen wieder zu glu abgebaut. in der leber hält die glykogenolyse den blutzuckerspiegel aufrecht, im muskel liefert sie glucose für den anaerobe glykolyse. 1) glykogen-phosphorylase ist das wichtigste abbauende enzym, es spaltet vom nicht reduzierenden ende her nacheinander glukose bausteine als glucose-1-phosphat ab. 2)Glu-1-ph wird zu glu-6-ph isomerisiert 3) umwandlung in freie glu |
| Hormone die in den Kohlenhydratstoffwechsel eingreifen ?? | Zu den Hormonen die in den Kohlenhydratstoffwechsel eingreifen gehören die peptide Insulin&glukagon, das glucocorticoid cortisol und das katecholamin adrenalin. |
| Leber-> Glukokinase besonderheit | bei niedrigenm blutzuckerspiegel ist glukokinase unwirksam, da sie durch translokation dann in den zellkern wandert. Das verstecken der glukokinase, verhindet, dass bei glukosemangel, durch glykogenabbau gebildete glucose intrazellulär in glu-6-ph umgewandelt wird und die zelle dann nicht mehr verlassen kann. |
| Glucosestoffwechsel der Leber, Leitstungen | Zu der wichtigsten leistung der leber gehört, die überschüssige glucose in form von glukagon zu speichern und daraus bei bedarf wieder glucose freizusetzten. sind die glykogenvorräte ausgeschöpft kann die leber gluconeogenese betreiben. außerdem baut sie wie alle anderen gwb. Glu über glykolyse ab. Abbau und aufbau finden natürlich nicht gleichzeitig statt, deshalb gibt es zwei schlüsselenzyme, die nur die anabole bzw. katabole reaktion katalysieren und unterschiedl. gesteuert werden. |
| cAMP wirkung?? | cAmP führt zu einer reversiblen phosphorylierung= interkonvertierung verschiedener enzyme! |
| Interkonvertierung? | ein und ausschalten von enzymen durch phosphorylierung, Diese modifikationen sind reversibel. phosphorylierung durch kinasen sind atp abhängig, unter wasserfreisetzung. phosphatasen= enzyme die phosphatgruppen hydrolytisch wieder von den proteinen abspalten können |
| Glukagon, Wirkung | Glykogen abbau!!!Glukagon signalisiert der leber den bedarf an glu im organismus.cAMPspiegel steigt(intrazellulärer botenstoff )in den zellen und die interkonvertierbaren enzyme also glykogenabbauende schlüsselenzyme werden phosphoryliert d.h.aktiviert GLYKOGENPHOSPHORYLASE [die glykogen abbauenden enzyme sind phosphoryliert aktiv] Glukagon:induziert enzme der gluconeogenese,hemmt die pyruvatkinase der glykolyse interkonversion von enzymen, werden durch cAMP vermittelt: auf diese weise wird die synthese von glykogen gehemmt, der glykogenabbau dagegebn aktiviert, sympathikusaktivierung, somatotropin, sinken der fettsäurekonz |
| Insulinwirkung | Glykogenaufbau!! Insulin senkt den cAMP spiegel in den zellen und sorgt für dephosphorylierung der entsprechenden enzyme. Glykogensynthase wird durch phosphatabspaltung aktiviert und baut glykogen auf[die glykogen aufbauenden enzyme sind dephosphoryliert aktiv] GLYKOGENSYNTHASE Insulin: aktiviert Glykogensynthase und aktiviert die schlüsselenzyme der glykolyse (hexokinase, 6-phosphofructokinase und pyruvatkinase), gleichzeitig uterdrückt es die synthese von enzymen der gluconeogenese |
| Insulinrezeptoren, eigenschaften und wirkungsweise | Insulinrez: Tyrosinkinase-Rezeptoren=tetrameres Protein, das die Zellmembran durchsetzt,aus je 2 α- und β-Untereinheiten, die jeweils durch Disulfidbrücken kovalent miteinander verbunden sind. Bei Bindung von Insulin an die α-UE>Konformationsänderung, welche die Kinase-Aktivität der β-Untereinheiten in Gang setzt. Es werden Tyrosinreste des Rezeptors phosphoryliert. >Eigenaktivierung des Rezeptors >Anlagerung eines intrazellulär gelegenen Signaltransduktionsproteins auch die Tyrosinreste dieses Proteins werden phosphoryliert >Signaltransduktion nach intrazellulär.Dabei werden auf verschiedenen Wegen die Transkription von Genen des Glucosestoffwechsels, die Zellproliferation, die Funktion der Phosphodiesterase und die Translokation von Glucosetransportern beeinflusst. |
| Insulin Regulatoren | Insulin freisetzung stimuli: Blutglu steigt, Ach(vagusstimulation, anreger der verdauung), hohe fettsäurespiegel im blut Hemmung durch: Hormone die den camp spiegel in bzellen erniedrigen: somatostatin, adrenalin noradr. !! |
| Cortisol wirkung auf kohlenhydratstoffwechsel? | Cortisol: induziert gluconeogenese und induziert enzyme des A.S. abbaus-> gluconeogenesevorstufen |
| von gierke speicherkrankheit? | Der Morbus von Gierke ist eine Glykogen-Speicherkrankheit, die durch einen Mangel des Enzyms Glukose-6-Phosphatase gekennzeichnet ist. Damit kann das beim Glykogenabbau anfallende Glukose-6-Phosphat nicht mehr dephosphoryliert werden und nicht die Leber- und Nierenzellen verlassen. Die Muskulatur, die das enzym ohne hin nicht besitzt ist von der krankheit nicht betroffen. Symptome dieser Erkrankung sind Hepatomegalie und später Nephromegalie, Hypoglykämie, Hyperlipämie, Hyperurikämie, Acetonämie und Hornhautdystrophie. Im weiteren Verlauf der Erkrankung treten hämorrhagische Diathesen und Leberinsuffizienz auf. Der autosomal-rezessiv vererbliche Morbus von Gierke ist mit Kleinwuchs und "Puppengesicht" assoziiert |
| Gluconeogenese: -Ausgangsstoffe -wo findet sie statt? | Die Gluconeogenese ist ein Stoffwechselweg zur Neusynthese von Glucose. Sie findet vorwiegend in der Leber und in den Nieren statt. Die wichtigsten ausgangsstoffe sind glucogene aminosäuren (vor allem alanin und glutamin, entstehen durch proteolyse im muskel) , lactat (wird in erys und unter o2 mangel im muskel gebildet) und glycerol (aus abbau von fett) die gluconeogenese kann auch ohne externe glucosezufuhr das gwb wochenlang versorgen!! |
| Wie viel Energie verbraucht die gluconeogenese? | Gluconeogenese verbraucht pro glu 4 ATP und 2GTP |
| Gluconeogenese ablauf | 1) Lactat- DH oxidiert lactat unter bildung von NADH zu pyruvat 2) pyruvat wird in die mitochondrien transportiert. dort über pyruvatcarboxylase zu oxalacetat carboxyliert (biotinabhgg reaktion)!!!! Das in den Mitochondrien gebildete Oxalacetat wird über reaktionen des malat-shuttle aus den mitoch. Exportiert 3)Im Zytolasma wandelt PEP-Carboxykinase, Oxalacetat unter verbrauch von GTP in Phosphoenolpyruvat um. 4) die weiteren schritte bis zu Fru-1,6- bisphosphat sind umkehrreaktionen der glykolyse 5) fru-1,6-bisphosphatase spaltet pho reste ab >fru-6-phosphat wichtiger regulationspunkt der gluconeogenese!! 6)Isomerisierung in glu-6-ph 7) glu-6-phosphatase als integrales membranprotein im inneren des gER lokalisiert(kommt in leber vor aber nicht in muskeln) glu kommt dann von ER wieder ins zytoplasma, von dort wird sie dann schließlich über GLUT2 ins blut abgegeben. |
| Oxalacetat | oxalacetat= zwischenprodukt des citratzyklus, alle aminosäuren deren abbau in den zyklus mündet oder pyruvat liefert= glucogene A.S.(mit 2 ausnahmen Leucin und Lysin, sind alle proteinogenen A.S. glucogen) |
| Pyruvatkinasereaktion | -pyruvatkinase spaltet das phosphatgruppe von phosphoenolpyruvat ab und überträgts auf ADP, wobei pyruvat entsteht und ATP! IRREV, exergon |
| was passiert mit pyruvat im aeroben Modus | pyruvat wird der pyruvatdehydrogenasereaktion zugeführt, weiter zu acetyl coa verstoffwechselt ->> atmungskette |
| Lactatdehydrogenase-reaktion | Pyruvat wird durch die LDH zu lactat reduziert, um NADH+H+ zum NAD+ zu oxidieren. |
| Lactat= endprodukt von?? was passiert damit? | Lactat ist das endprodukt der anaeroben glykolyse und kann über blutweg der leber zugeführt werden, in der leber wirds für gluconeogenese verwendet, wobei die entstehende glu den zellen wieder für glykolyse zur verfügung steht= CORIZYKLUS |
| Pentosephosphatweg, Funktion und Produkte? | Nebenweg der Glycolyse. Er erzeugt NADPH für anabole stoffwechselwege (biosynthese von fettsäuren, und isoprenide) Sowie Pentosen (Biosynthese von Ribose-5-phosphat) für die Nukleotid Biosynthese (Purin- und Pyrimidin- einschl. Purin-Salvage) oxidativer stoffwechselweg im zytoplasma,weniger als 10% des glu-6-ph werden über pphoweg abgebaut |
| Ablauf des PPW und Nukleotidsynthese? | glu-6-ph wird in ribulose-5-pho umgewandelt, dabei werden 1 co2 und 2 nadph gebildet. 1) glucose-6-ph-dehydrogenase oxidiert glu-6-ph zu 6-phosphogluconlacton, dabei wird das erste nadph gebildet 2)spezifische hydrolase spaltet das lacton und legt die carboxygruppe des 6-phosphogluconats frei 3)phosphogluconat-dehydrogenase, spaltet carboxylatgruppe des 6-phosphogluconats als co2 ab und oxidiert gleichzeitig an c3. Das zweite nadph und ribulose-5-pho (ketopentose) entsteht, durhc eine isomerase wird dadurch ribose-5-pho, die ausgangsverbindung der nukleotidsynthese!!! der regenerative teil des weges, wandelt pentosen wieder in hexosen um, wenn keine pentosen benötigt werden aber nadph benötigt wird. Und schleust hexosen wieder in die glykolyse ein. nukleotidsynthese : Phosphorylierung von alfa-D-ribose-5-phosphat am ersten kohlenstoffatom! PRPP entsteht |
| Was ist die elektrophorese? | Die elektrophorese ist eine methode um verschiede proteine und A.S. voneinander zu trennen. Diese methode wird genutzt um die zusammensetzung der plasmaproteine festzustellen, bei diversen krankheiten kann die konz. des einen oder anderen proteins erhöht/erniedrigt sein. |
| Ablauf einer elektrophorese, wie funktioniert die aufteilung in fraktionen? | Dazu trägt man ein proteingemisch also patientenserum auf ein gel auf und legt eine spannung an, worauf die proteine anfangen zu wandern. Die meisten proteine wandern zum pluspol. Wanderungsgeschwindigkeit hängt von größe und ladung der einzelnen proteine ab. Danach legt man den trägerstreifen in ein färbebad, mit dem die einzelnen proteinbanden dargestellt werden können. Die konzentrationen werden photometr. Gemessen und graphisch dargestellt. Serumfraktionen: Albumin 60%, alfa1globulin 4%, alfa2g 8%, beta-g 12%, gamma –g 16% durch die prozentzahlen lassen sich ne menge infos ableiten. |
| Plasmaproteine: -Konz im Blutplasma? -Wo gebildet? | Plasmaproteine im blutplasma, haben ne gesamtkonz. Von 7,5g/dl Alle plasmaproteine sind glykoproteine bis auf albumin, -Leber =hauptproduzent der pproteine, funktionseinschränkung der leber in erster linie bemerkbar durch verrinerung der albuminfraktion |
| alfa, beta und gamma globuline | -gamma-gl.sind immunglobuline=antikörper und werden durch plasmazellen produziert, ihre verminderung= eingeschränkte bildung von imunglobulinen -beta-gl.= fibrinogen(gerrinungssystem) und transferrin(transportiert eisen-ionen) -Zu den Alpha-1-Globulinen gehören die so genannten Akutphasenproteine, also Proteine, die in der akuten Phase einer Entzündung erhöht sind. Zu einer Erhöhung der alpha-1-Globuline kann es daher bei akuten Entzündungen verschiedener Ursache kommen |
| Translation: wo findet sie statt? | die aktivierung der A.S. findet im zytosol statt, danach: mRNA gelangt durch die kernporen ausm zellkern ins zytosol, wird dort vonnem ribosom gebunden, dort erfolgt die translation |
| Translation was passiert dort? grobe funktion | die translation wird nur von mRNA bestritten. Die info die auf der mrna steht, dient als vorlage für die proteinsynthese. Mrna besteht aus nukleotiden. Diese nukleotide müssen in aminosäuren übersetzt werden= translation/Übersetzung>> dabei werden nach dem plan der mrna einzelne A.S. zu nem protein aneinandergereiht |
| Aktivierung der A.S. für Translation? | 1) aktivierung der aminosäuren: zur aktivierung wird jede A.S. an tRNA gebunden, hierbei entsteht ne energiereiche esterbindung (bei deren spaltung genug energie frei wird, um A.S. zu nem peptid zu verknüpfen. 3Basen der mRNA=codon >>kodieren für eine bestimmte A.S. die tRNA, die die betreffende Aminosäure trägt, hat am Anticodon Arm die 3 zum Codon der mRNA KOMPLEMENTÄREN BASEN! Die tRNA bindet mit dem anticodon arm ans basentriplett der mrna, an ihrm 3‘ ende trägt die tRNA die nukleotidsequenz CCA an welche die A.S. gebunden ist, d.h. A.S. ist immer am Adenosin der tRNA gebunden. tRNA ist also übersetzer zw. mRNA und dem entstehenden peptid. tRNA besteht aus 80nukleotiden. |
| Was ist die aufgabe von tRNA? | tRNA ist also übersetzer zw. mRNA und dem entstehenden peptid. tRNA besteht aus 80nukleotiden. die tRNA, die die betreffende Aminosäure trägt, hat am Anticodon Arm die 3 zum Codon der mRNA KOMPLEMENTÄREN BASEN! Die tRNA bindet mit dem anticodon arm ans basentriplett der mrna, |
| wieviele tRNAs gibt es, wieviele Aminosäuren? | Wobble-Hypothese: Es gibt 61 versch. Codons die für ne A.S. codieren aber nur 31tRNAs (in mitos nur 22) bei basenpaarungen ist das dritte nukleotid nicht so wichtig, und daher sind leicht veränderte paarungen mögl. |
| Was sind Codons? | 3 aufeinanderfolgende basen eines strangs also ein basentripplett kodiert genau für eine aminosäure. codon ist die grundeinheit des genetischen codes. Bei ner kombination von 3 basen erhältman 4hoch3 also 64 unterschiedl. codons, genug um die information für die 20 aminosäuren zu speichern. 64 codierungsmöglichkeiten aber nur 20 A.S. sodass einige A.S. mehrfach kodiert werden-> degenerierter genetische code, aber bei den meisten A.S. sieht man schon an den ersten 2 buchstaben welche es ist z.b. GG->glycin |
| Codesonne? | die verschiedenen basen stellen das alphabet unseres genoms dar, eine übersichtliche darstellung aller mögl. Basentripplets erreicht man mit der codesonne gelesen wird sie strahlenförmig von innen nach außen. die codesonne bildet den schlüssel zur übersetzung der basensequenz auf der dann in eine abfolge von A.S. eines proteins Nur 61 der 64 tripletts codieren für A.S., es gibt näml. 3 stopcodons, die das ende eines gens signalisieren, also zum kettenabbruch führen. UAA,UAG,UGA |
| Besondertheit des Trippletts AUG? | vereint 2 informationen in sich! Codiert einmal für A.S. methionin und ist das einizge Startcodon-> signalisiert beginn eines gens |
| Translationsinitiation | Translationsinitiation-zsmbau der ribosomen Translation erfolgt am ribosom also einem rRNA-Komplex und verschiedenen Proteinen. die beiden UE 60s(3rrnas),40s des ribosoms liegen im zytosol getrennt und müssen erstmal mit hilfe von initiationsfaktoren zsmgebaut werden= 80s startcodon AUG auf mRNA kodiert für methionin>> immer 1.A.S. des proteins! |
| Translationselongation | kleine RibosomenUE hat 2 bindungsstellen für tRNAs, eine mit methionin beladene tRNA=starter tRNA wird an die Peptidyl bindungstelle gebunden. die tRNA deren anticodon zum nächsten codon auf der mrna passt, bindet an der Aminoacyl bindungstelle hier helfen elongationsfaktoren den beladenen tRNAs beim binden, die aminosäuren verbinden sich durch peptidbindungen. die entladene tRNA wird abgespalten und das ribosom wandert auf der mrna um ein basentriplett in richtung 3‘ ende der mRNA weiter. |
| Translation Termination: | Termination: taucht an der aminoacylbindungsstelle einer der 3 stopcodons: UAG,UAA,UGA auf, bindet statt ner beladenen tRNA ein FREISETZUNGSFAKTOR. Es bewirkt dass die fertige peptidkette durch ein wassermolekül abgespalten und freigesetzt wird. daraufhin zerfällt das ribosom wieder in seine UE |
| Wie wissen frisch synthetisierte proteine wo sie hin müssen? | Proteinsortierung! Um ihren weg zu finden tragen die proteine signalsequenzen, die von rezeptorn der transporter gelesen werden, proteine ohne signal bleiben nach synthese im zytosol. |
| Erklären sie wie eine zum Export bestimmte Polypeptidkette zum rER dirigiert wird; was passiert mit der Signalsequenz? | Sekretorischer Weg: Proteine am N-terminus mit signalpeptid für rER ausgestattet, sobald das signalpeptid auf dem ribosom erscheint, bindet ein rna haltiges signalerkennung partikel (SRP) an die sequenz und unterbricht die translation. Das SRP bindet dann an einen srp rezeptor in der membran des rER und fixiert das ribosom am rER. Danach löst sich SRP vom rezeptor ab. Die Signalsequenz ist immernoch im ribosom enthalten. Nun wird das protein nach und nach ins rER lumen eingeführt, dort spaltet noch während der translation eine signalpeptidase der inneren rER membran die signalsequenz ab. nach posttranslationalen modifizierungen im rer, golgi entsteht das reife protein. |
| Translation Welche Proteine müssen ins ER? | Sekretorische proteine/exportproteine, membranproteine,proteine für er und golgi, lysosomale proteine(da die lysosomen vom golgi gebildet werden) |
| PosttranslationaleProzessierung | die zelle muss ne reihe kotranslationale und posttr. Modifizierungen vornehmen, damit die proteine biolog. Aktiv werden können und ihre fkt. Wahrnehmen können. 1.zunächst müssen sich die proteine nach der translation richtig falten 2.dann erfolgen modifizierungen der proteine vor allem die glykosylierung |
| Proteinfaltung | Chaperone= wichtigste katalysatoren der proteinfaltung-> wichtige rolle spielen dabei hitzeschockproteine wenn proteine durch membranen wollen, müssen sie dafür entfaltet werden und nach passage wieder zurückgefaltet werden>> auch hier übernehmen hitzeschock-proteine die faltung |
| Glykolysierung | findet im rER und im golgi statt (intrazelluläre nicht glykolysiert, aber sekretor. Glykolysiert!) Anheftung einer kohlenhydratkette ans protein. die glykolysierung erfolgt an den A.S. serin, threonin (oh gruppe)und asparagin (NH gruppe) O glykosylierung: nur im golgi N glykosylierung:spezifisch im golgi und im ER, alle kohlenhydrate müssen zuvor mit UTP aktiviert werden |
| N glykosylierung Ablauf, an welche A.S.? | dolicholphosphat in ER membran, sachharidkette wird erst an dolicholpho syntetisiert. die sachharidkette am dolicholpyrophosphat zeigt dann ins ER lumen, weitere kohlenhydrate werden angelagert. gesamte kohlenhydratkette wird mithilfe einer bestimmten erkennungssequenz auf den asparaginrest des proteins übertragen. Dieses protein befindet sich gerade im ER lumen inmitten der translation=kotranslationale glykosylierung protein mit der kohlenhydratkette wird dann getrimmt , wandert dann im vesikel zu golgi app. im golgi werden die für das jeweilige glykoprotein spezif. Saccharidreste angeheftet (vorallem N-acetyl-glucosamin,galaktose,fukose,sialinsäure) |
| O glykolysierung, anheftung an welche A.S? | O: bekommen ihre zuckeranteile in zisternen des golgi app., anheftung des ersten zuckers an serin/threoninrest direkt an protein |
| beispiele für glykolysierte proteine? | >>glykoproteine >>blutgruppen antigene unterscheiden sich durch die glykosylierung der proteine auf eryoberfl. |
| andere Posttranslationale Modifikationen: | -Phosphorylierungen>> beeinflussung der biolog. Aktivität der enzyme, durch second Messenger cAmP führt zu einer reversiblen phosphorylierung= interkonvertierung verschiedener enzyme phosphorylierung durch kinasen sind atp abhängig, unter wasserfreisetzung. -acetylierung am n-terminus |
| Abbau von PROTEINEN, welche Enzyme sind dafür verantwortlich? | proteinabbauende enzyme=peptidasen(gehörn zu hydrolasen, spalten peptidbindungen unter wasseranlagerung) Peptidasenarten: 1) verdauungsenzyme unspezifisch 2)extrazelluläre peptidasen(mit spezif. Fktionen) 3)intrazelluläre peptidasen (z.b. in lysosomen) exopeptidasen= spalten am c oder n terminus, substratspezifisch endopeptidasen=proteinasen weil sie protein iwo in der mitte spalten, nicht subspez proteine werden ausschließlich innerhalb der zellen abgebaut, daneben gibt’s noch spezialisierte zellen die ganz bestimmte proteine abbauen |
| Abbau von zytosolischen Proteinen im 26S-Proteasom: | ATP abhängig ein proteasom ist ein multienzymkomplex, im zytosol. Eine unspezifische protease. abgebaut werden nur protein, die zuvor mit mehreren ubiquitinen markiert wurden ubiquitinierung erfolgt an der A.S. Lysin des abzubauenden proteins, ubiquitin wird bei proteinabbau, freigesetzt und wiederverwendet. |
| Proteinabbau im Lysosom , ihre Enzyme? | baun keine einzelnen proteine sodern gleich ganze organellen ab, sie besitzen eine ganze reihe von hydrolysen wie peptidasen, diese trennen die peptidbindungen auf ATP getriebene protonenpumpen in der lysosomalen membran-> saures milieu im lysosom von ph=5 Leitenzym: saure phosphatase, andere enzyme: glykosidasen, lipase, protease, nuklease, lysozym Aufgaben des Lysosoms: AUTOPHAGIE können entweder eigene stoffe abbauen HETEROPHAGIE oder abzubauendes fremdmaterial, z.b. in den körper gedrungene bakterien, extrazelluläre stroffe werden per endozytose o. phagozytose aufgenommen und abgebaut, dabei verschmilzt das endosom mit dem lysosom zum sekundären lysosom |
| Peptidbindung zeichnen können | Bindung zwischen 2 A.S.=peptidbindung peptidbindung: wenn die aminogruppe einer A.S. mit der carboxylgruppe der anderen A.S. reagiert, dabei wird wasser abgespalten. CO-NH bezeichnet man als säureamid also peptdbindung ist eine säureamidbindung Bild: Kondensation einer Carboxy- und einer Amino-Gruppe unter Wasser-Abspaltung |
| Proteine: Wie sind Proteine aufgebaut (Primär-, Sekundärstruktur etc., Helices und Faltblätter erklären)? | Die struktur eines proteins wird auch als konformation bezeichnet Primärstruktur: abfolge der einzelnen A.S. innerhalb der kette Sekundärstruktur: alfaHelix schraubenförmig gewundene anordnung der polypkette, dabei gehen die CO und NH gruppen miteinander wasserstoffbrückenbindungen ein beta Faltblatt peptidkette liegt in zickzackform vor, hier können sich wasserstoffbrücken intra du intermolekular bilden (zwischen 2 proteinen) Tertiärstruktur: beschreibt die 3d struktur des proteins und entsteht durch verwindung der sekundärstruktur, seitenketten der A.S.gehen bindungen miteinander ein, je nach seitenketten, disulfidbrücken, wasserstoffbbindungen, hydrophobe wechselbwirkungen hydrophile gruppen zeigen nach außen-> hydrathülle Quartärstruktur:supramolekulare strukturen =proteinuntereinheiten, mehrere tertiärstrukturen schließen sich zu funktionsUE zsm, z.b. ribosom |
| POLYMERASE-KETTENREAKTION Wozu? | Vervielfältigen von DNA, als diagnostischer nachweiß |
| was man alles für eine PCR braucht?? | -Original-DNA, die den zu vervielfältigenden Abschnitt enthält -Man benötigt oligos, das sind 2 spezif. Primer(DNA einzelstrangstücke mit 20nukleotiden) die bestimmen welcher abschnitt amplifiziert werden soll -nukleotide in desoxyformen TaqPolymerase: muss hitzestabil sein bis zu 90°C Puffer: dna polymerase arbeitet bei bestimmtem ph wert, deshalb brauch man nen puffer der den ph hält Mg2+-Ionen, für die Funktion der Polymerase essentiell, stabilisieren die Anlagerung der Primer und bilden lösliche Komplexe mit den Desoxyribonucleosidtriphosphaten |
| In welchem Gerät findet die PCR statt? Und wie viele Zyklen sind nötig für den PCR prozess? | Die Polymerase-Kettenreaktion findet in einem sogenannten Thermocycler statt. Diese Maschine erhitzt und kühlt die in ihr befindlichen Reaktionsgefäße präzise auf die Temperatur, die für den jeweiligen Schritt benötigt wird Der PCR-Prozess besteht aus etwa 20-50 Zyklen. Jeder Zyklus besteht aus drei Schritten |
| PCR Ablauf,die 3 schritte | 1. Denaturierung: Zunächst wird die doppelsträngige DNA auf 95 °C erhitzt, um die Stränge zu trennen. Die Wasserstoffbrückenbindungen, die die beiden DNA-Stränge zusammenhalten, werden aufgebrochen. Danach wird schnell auf 65 °C abgekühlt, um die Rückbildung der Doppelhelix zu verhindern. 2.Primerhybridisierung: Temperatur wird30s lang auf einem Wert gehalten, der eine Anlagerung der Primer an die DNA erlaubt. Die genaue Temperatur wird hierbei durch die Länge und die Sequenz der Primer bestimmt Meistens 55-65° 3.Elongation:Schließlich füllt die DNA-Polymerase die fehlenden Stränge mit freien Nukleotiden auf. Sie beginnt am 3'-Ende des angelagerten Primers und folgt dann dem DNA-Strang. Der Primer wird nicht wieder abgelöst, er bildet den Anfang des neuen Einzelstrangs. 70°C. Dieser Schritt dauert etwa 30 Sekunden je 500 Basenpaare |
| PCR Anwendung in der Klinik? Primer?? | Vaterschaftstest, VirusDNA im blut nachweisen, Erstellung eines genetischen Fingerabdrucks, Man benötigt wie bei der dna-replikation, spezif. Primer als startpunkte für die dnapolymerase. verwendet man primer, die spezifisch an ein bestimmtes virusgenom binden können, so funktioniert die PCR nur dann, wenn dieses virus auch im ansatz da war, also wenn patient infiziert ist. Im anderen fall entsteht kein pcr produkt. primer, die an HIV genom binden sollen-> HIV test |
| Unterschiede von In-vivo- und In-vitro-DNA-Synthese | -wie bei der replikation synthetisiert bei der PCR eine DNApolymerse den komplementären strang zum gewünschten DNAabschnitt aber durch eine mehrmalige wiederholung erhält man eine hohe vervielfältigungsrate, es werden immerwieder kopien von kopien erstellt, deshalb spricht man von kettenreaktion. -Bei einer Standard-PCR können dies bis zu etwa dreitausend Basenpaare (3 kbp) lange DNA-Fragmente sein. Eine menschliche Zelle enthält beispielsweise etwa drei Milliarden Basenpaare pro haploidem Genom. |
| Warum macht man bei einem Neugeborenen einer HIV-positiven Mutter keinen Antikörpertest mittels ELISA, sondern eine PCR? | Elisa: Prinzip dieses Tests ist es gegen HIV gebildete Antikörper im Blut nachzuweisen, er weist aber nicht das Virus selbst nach!! Sondern nur ob Antikörper gegen die viren vorhnden sind. weil der Antikörpernachweis falsch positiv ausfallen könnte, übertragene IgG-AK der Mutter sind plazentagängig) |
| Citratzyklus: -was geht rein | Acetyl CoA (Abbauprodukte der 3 nährstoffe) |
| Citratzyklus, was kommt raus ? bilanz | der citratzyklus ist ein kreisprozess da nach den 8 reaktionen wieder oxalacetat vorliegt Bilanz: 2moleküle co2, 1 GTP, NADH/H+ und FADH2 reduzierte coenzyme |
| wie wird Pyruvat zu Acetyl-CoA?? | Durch die Pyruvat Dehydrogenasereaktion wird Pyruvat zu mitoch. Gebracht und dort zu AcetylCoA umgewandelt. Dehydrierende(oxidative) Decarboxylierung also wasserstoff und co2 werden gleichzeitig abgespalten! Oxidation durch Liponamid, Decarboxylierung durch Tiaminpyrophosphat IRREV aus fettsäuren die zu acetylcoa abgebaut werden kann niemals glukose werden! Weil acetyl coa nicht in pyruvat umgewandelt werden kann! PDH= mutlienzymkomle aus 3 verschiedenen enzymkomplexen, durch die umwandlung von pyruvat in acetylcoa verbindet die Pyruvatdehydrogenase die glykolyse mit dem citratzyklus und leitet damit den aeroben abbau der glucose ein! PDH: Pyruvat+CoA+NAD+-> acetyl CoA+CO2+NADH+H+ Regulation der PDH? profukthemmung und interkonvertierung-> in phosphoryliertem zustand ist PDH komplex inaktiv (phosphoryliert durch PDH kinase) |
| citratzyklus: INPUT and OUTPUT | Input Acetyl Co (entsteht beim Glu abbau, fettsäure abbau und einige A.S) nichtred. Coenzyme nad fad kohlenstoffgerüst der A.S. Output NADH+H+ und FADH2; CO2, GTP, Oxalacetat(glucose aufbau), Succinyl-CoA (für porphyrinbiosynthese) ES WIRD KEIN ATP GEBILDET NUR GTP!! |
| Citratzyklus, wichtigste funktion | citratzyklus: =amphiboles zentrum wichtigste funktion ist die umwandlung von acetyl coa zu 2CO2! Die dabei frei werdende energie wird als NADH+H+ und FADH2 fixiert [konservierung der energie die beim acetylcoa frei wird!] und bei der atmungskette verwendet. enzyme des cz befinden sich im mitochondrium, genau wie atmungskette>> leichter weg für die reduzierten coenzyme |
| Citratzyklus schritte/ablauf kurz | Citratsynthase: übertragung von acetylcoa auf oxalacetat unter bildung von citrat Citrat-> acotinase-> isocitrat Isocitrat DH-> alfa ketoglutarat alfa ketoglutarat DH-> succinyl coa Succynyl coa syntetase >succinat > fumarat-> malat-> oxalacetat |
| Citratzyklus -welche Stoffe kann der Körper sich für andere Stoffwechselwege abzweigen? Intermediate?? | A.S Biosynthese: Oxalacetat-> Aspartat->Asparagin, alfaketoglutarat-> Glutamat Glukoneogenese: Oxalacetat-> PEP carboxykinase-> PEPyruvat Fettsäure Biosynth: benötigt Acetyl coa-> erstmal reaktion mit oxalacetat zu citrat um membran zu durchdringen und ins zytosol zu gelangen HämBiosynth: Succinyl CoA=ausgangsstoff Intermediate, die für den anabolen stoffwechsel ausm zyklus entfernt werden, müssen natürlich ersetzt werden durch>> anaplerotischen reaktionen =auffüllreaktionen, die zur bildung dieser intermediate führen (bsp. Bildung von oxalacetat aus pyruvat durch PYRUVATCARBOXYLASE) |
| Laktoseintoleranz? Warum Blähungen? | genetische Information über Laktase fehlt wenn die laktaseaktivität fehlt, fehlt auch der blutglukoseanstieg, der gemessen werden kann. die ins kolon gelangende laktose wird durch die darmflora zersetzt, es entstehn organische säuren udn gase, die blähungen und durchfälle verursachen. |
| Aktivierung der Thrombozyten: | Adhäsion der thrombos an die EZM führt zur ausschüttung granulärer inhaltsstoffe-> führn zu einer endgültigen irrev. Aktivierung der thromb. Ausbildung von pseudopodien-> verschießen die wunde |
| vWF Rolle während Hamöstase? Was bewirkt es? | vWF ist ein Hilfsprotein bei der thrombozytenadhäsion, vWF wird von endothelzellen hergestellt. Im plasma liegen vWF multimere zsm mit dem gerinnungsfaktor8 vor und in den thromboz. Befindet sich auch ein wenig vWF Bindung des vWF an Kollagen, kollagen entfaltet sich und es entstehen bindungsstellen für thrombozyten, diese können so nicht nur auf kllg direkt sondern auch via vWF an die subendotheliale matrix gebunden werden. ALSO VWF=BRÜCKE ZW SUBENDOTHELIALER MATRIX UND THROMBO Die freigelegten bindungsstellen dienen als liganden für den thrombozytären vWF-Rezeptor= GP1b/9 Komplex, ein Integrin |
| Granula der Thrombos: | dichte granula: sind die ersten, die nach thromboaktivierung ausgeschüttet werden, enthalten ADP und z.b. serotonin, das weitere thrombos aktiviert. Und Ca2+-> für die weitere gerinnung notwendig ADP aktiviert den GP2b/3a Rez.= fibrinogenrez. Der entscheidend ist für thrombozytenaggregation Thromboxan: wirkt ebenfalls aktivierend auf andere thrombos, führt zur gefäßkonstriktion alfa Granula: nur proteine-> fibrinogen, gerinnungsfaktoren, vWF (wichtig für adhäsion und aggregation) lysosomale granula:enthalten hydrolyt. enzyme Der GP2b/3a Rez: ADP wird ausgeschüttet und bindet an ADP rez auf dem gleichen thromozyten aber auch auf nachbar thromboz. durch die anschließende signaltransduktionskaskase kommts zu ner konformationsänderung am GP2b/3a rez, der fibrinogen bindet, auf diese weise werden 2 thrombos miteinander verbunden, über fibrinogen |
| Vitamin K Abhängigkeit von gerinnungsfaktoren | faktor 10 7 9 2 werden nach ihrer biosnthese in der leber noch modifiziert, mit hilfe vit K erfolgt posttranslationale gamma-carboxylierung von glutamatresten, erst durch diese modifikation wird die calcium vermittelte bindung an de neg. geladenen phospholipidoberfl. Möglich |
| Fibrinolyse? | entfernen von blutgerinnseln, plasmin=zentralenzym aktivierung streng reguliert durch plasminogen-aktivatoren(t-PA ein glykoprotein aus den endothelz.) plasmin wird in der leber hergestellt Arbeitsweise: plasmin bindet an fibrin, und zerschneidet es in d-dimere und e-fragmente, t-PA spaltet neben fibrin auch noch fibrinogen und vWF |
| Regulation der hämostase | Cyclooxygenase stellt in den endothelzellen aus arachidonsäure Prostazyklin her. Prostazyklin wird ans blut abgegeben, thrombozyten besitzten rez für prostazyklin, bei ligandenkontakt->intrazelluläre cAMP erhöhung, hemmung der thrombozytenaggregation wenn endothelzellen verschwinden, fällt diese hemmung auch weg Gegenspieler von Prostazyklin= Thromboxan in den thrombozyten thromboxan ist thrombozyten aktivierend und aggregierend |
| ASS, CUMARIN, HEPARIN WIRKING AUF HÄMOSTASE | ass Wirkung: hemmt cyclooxygenase also hemmt produktion von prostazyklin und von thromboxan hemmt cyclooxygenase IRREV! in den endothelz, wird cox recht schnell nachsynthetisiert, also hemmung ds prostazyklins nur begrenzt! thromboxanhemmung dauert viel länger, es müssen erstmal neue thrombozyten gebildet werden, damit neue cycooxygenase entsteht Cumarin= vit K antagonist!-> hemmt blutgerinnung! Herstellung fehlerhafter gerinnungsfaktoren in der leber, diese werden nicht modifiziert! heparin= antikoagulatie, hemmt faktor 10 und thrombin, aktivator der fibrinolyse |
| Transendotheliale Migration LEUKOZYTENMIGRATION? | Als transendotheliale Migration bezeichnet man die Wanderung (Migration) bestimmter, amöboid beweglicher Zellen (z.B. Leukozyten) durch die Gefäßwand der Kapillaren (Endothel). Die transendotheliale Migration ermöglicht den Zellen des Immunsystems das Gefäßsystem zu verlassen, um beispielsweise an einen Infektionsort zu gelangen und dort den Entzündungsprozess zu unterhalten. Den Prozess des "Anlockens" der Zellen nennt man Chemotaxis. • Zunächst vermitteln lektinartigen Adhäsionsmoleküle, die Selektine, das "Andocken" der Leukozyten auf der Endotheloberfläche. • Die Leukozyten werden verlangsamt und treten in Kontakt mit speziellen Zytokinen auf der Endotheloberfläche auf, die man als Chemokine bezeichnet. • Die Chemokine stimulieren wiederum die Aktivierung von Integrinen auf der Leukozytenoberfläche. Sie vermitteln die Bindung der Zellen an das Endothel. • Die stabil gebundenen Leukozyten können sich gezielt fortbewegen und durchwandern schließlich aktiv die Endothelzellschicht. |
| Gluthation, was ist es? | Glutathion ist ein atypisches Tripeptid aus den Aminosäuren Glutamat, Cystein und Glycin. Atypisch ist die Bindung deshalb, weil sie unabhängig vom Proteinbiosyntheseapparat erfolgt und die γ-Carboxylgruppe des Glutamats die Peptidbindung eingeht. Katalysiert wird die Bildung durch die Glutathionsynthetase unter Verbrauch von ATP. Glutathion kommt in sämtlichen Körperzellen vor, hat eine herausragende Bedeutung aber vor allem in den Erythrozyten. |
| Gluthation, biolog. funktion | Glutathion sorgt für die Aufrechterhaltung reduzierender Bedingungen in den Körperzellen, d.h. es reduziert reaktive Sauerstoffverbindungen und schützt so die Zelle vor Schäden durch Radikale. Besonders wichtig ist seine Anwesenheit in den Erythrozyten: hier reduziert es Methämoglobin, welches keinen Sauerstoff mehr transportieren kann, zu funktionsfähigem Hämoglobin!! Wichtig für seine Funktion ist die Sulfhydrylgruppe (-SH) des Cysteins, die bei der Reduktion von Wasserstoffperoxid mittels der Glutathion-Peroxidase oxidiert wird und sich mit der analogen SH-Gruppe eines weiteren Glutathions zu einer Disulfidbrücke verbindet: 2 GSH --> GSSG Zur Regeneration dient die Glutathionreduktase, welche die Disulfidbrücke NADPH-abhängig reduziert. Das erforderliche NADPH stammt aus dem Pentosephosphatweg. |
| Diabetes mellitus? Folgen? | ist eine stoffwechselkrankheit die auf absoluten oder relativen mangel an insulin zurückgeht. Das fehlen des insulins wirkst sich auf stoffwechsel der kohlenhydrate und lipide aus. Typ1 Bzellen werden durch autoimmunreaktion in frühem alter zerstört Typ2 setzt in höherem alter ein, insulin resistenz entwickelt sich Auswirkungen eines Insulinmangels: Lipidstoffwechsel wird beeinflusst, insulin fördert umbau von glucose in fettsäuren>>aktivierung der acetyl-coa-carboxylase Hyperglykämie: glu spiegel im blut , glu aufnahme und verwertung in musklz. Und fettz. Beeinträchtigt Gluconeogenese wird gesteigert, durch proteolyse in der muskulatur Glukosurie: wenn kapazität zur glu rückresorpion in der niere ausgeschöpft ist Insulinmangel steigert fettabbau, die anfallenden fettsäuren werden in der leber zur synthese von lipoproteinen verwendet-> hyperlipidämie rest wird zu acetylcoa abgebaut-> zu viel für citratz, überschuss an ketonkörper in der leber Glycierung: angiopathien , schädigung der nieren,neuropathien, linsentrübung |
| Insulinwirkungen | Wirkung des Insulins: förderung der glucoseverwertung und hemmung der gluconeogenese, Transport von glu von blut in gewebe (GLUT4= insulinabhängig) ausnahme leber,erys, zns |
| Ikterus: prä,post,intra | prähepatisch: Leber ist damit überfordert bilirubin abzubauen und auscheiden zu können. Das kann z.b. bei einer hämolyse der fall sein, wenn sehr viel bilirubin aus dem anfallenden hämoglobin entsteht Unkonjugiertes Bilirubin ist die nicht wasserlösliche Form des Bilirubins, die im Serum an Albumin gekoppelt transportiert wird. Durch Konjugation mit Glucuronsäure entsteht in der Leber das wasserlösliche konjugierte ("direkte") Bilirubin. intrahepatisch: infolge einer leberzirrhose können sich die kontakte zwischen leberzellen lösen, sodass galle zwischen freie laufbahn in die sinus hat und ins blut gelangen kann posthepatich: störung im bereich der extrahepat. Gallenwege, wenn diese z.b. durch gallensteine verschlossen sind, ergibt sich die extrahepatische cholestase und posthepatischer ikterus |
| Erythrozyt: einen Aufmalen | bikonkave, abgeplattete Form Durchmesser7,5 µm Dicke 2 µm Energiegewinnüber anaerobe Glykolyse. Hauptbestandteil der Erythrozyten ist das Protein Hämoglobin, das ihnen ihre charakteristische rote Farbe verleiht und für den Sauerstofftransport verantwortlich ist. Die Hämoglobinmenge im Zytosol eines einzelnen Erythrozyten beträgt 30pikogramm. |
| Die spezielle Form der Erythrozyten, verformbarkeit | Form führt zu gr.Oberfläche der erys und verbessert dadurch den Gasaustausch. Zusätzlich wird die Flexibilität verbessert, was den Erythrozyten ermöglicht, durch Kapillaren zu wandern, deren Durchmesser kleiner ist als sie selbstEin unter der Zellmembran befindliches und in diese einstrahlendes dichtes strukturgebendes Netz von Filamenten, das erythozytäre Zytoskelett, ermöglicht das Aufrechterhalten der bikonkaven Form. Einige Proteine, etwa Spektrin oder Ankyrin, sind für die Struktur essentiell |
| MCH normalwert? | = 30pg mittlere Hb gehalt eines Erys Hb bei Frauen 14g/dl bei Männern 16g/dl |
| Wie ist Hämoglobin aufgebaut? Bestandteile? | Hb besteht aus 4 UE, jede UE setzt sich aus einem Porphyrinteil(Häm-molekül) und einem Proteinteil(globin) zsm. Häm Molekül: Chelatbildung mit Fe2+, Ring aus 4 pyrrolringen=das porphyrinogen Das zentrale eisen-ion besitzt 6 koordinationsstellen: 4 davon werden von stickstoff atomen der pyrrolringe besetzt, eins dient der verbindung des häms mit dem globin(also mit dem histidinrest), und eins für den sauerstoff! GlobinMolekül: der proteinteil=globin= eine polypeptidkette, eine UE des Hb besitzt jeweils eine Polypeptidkette also ein globin. 1 Hb molekül enthält somit 4 peptidketten, davon sind 2 immer alfa ketten und die anderen beiden zwei sind auch vom gleichen typ HbA1: 98% des hbs eines erwachsenen, 2alfa und 2beta ketten HbA2 2% 2 alfa und 2delta ketten Fetales Hb aus 2alfa und 2 gamma ketten, fetales hb hat eine höhere sauerstoffaffinität |
| Hb synthese | Hb wird im knochenmark von den ery vorläuferzellen gebildet, wo sie noch organellen besitzen. Aus Succinyl CoA[von citratz.]+ Glycin entsteht durch Vit B6 die delta-Aminolävulinsäure Porphobilinogen-> protoporphyrin-> einbau des eisenions-> häm Häm wird im zytosol mit globin verbunden |
| Sauerstoffbindung von Hb und Bohreffekt? | O2-bindung= Oxygenierung!: hauptaufgabe des Hb, das gesamte Hb kann 4 O2 moleküle transportieren, aufbnahme von sauerstoff ist reversibel, bei hohem partialdruck in der lunge nimmt Hb sauerstoff auf, bei niedrigem(im gewebe) gibt es ihn wieder ab. Bei bindung eines sauerstoffmoleküls kommts zur konformationsänderung im Hb, sodass das nächste O2 molekül leichter binden kann-> O2affinität steigt! Sauerstoffbindungskurve: rechtsverschiebung-> abnahme der O2aff, bereit zur abgabe von sauerstoff an gwb, z.b. an arbeitende muskulatur ph=gering,CO2hoch linksverschiebung-> zunahme der O2aff Einfluss von Co2 und PH auf die sauerstoffaff= bohreffekt! |
| Co2Transport im Blut | Co2Transport im Blut 80% werden als bikarbonat im blut transportiert 10% in ery an hb gebunden 10% direkt im blut gelöst Bikarbonat entsteht im ery aus kohlensäure, Bikarbonat diffundiert aus Ery hinaus ins blut, im austausch von Cl- Cl- verschiebung von Blutplasma in Ery= Hamburgershift! Im blutplasma wird bikarbonat zur lunge transportiert, dort muss CO2 abgeatmet werden, sodass bikarbonat wieder mittelc cl- in ery diffundiert und dort zu CO2 umgewandelt wird |
| Hb als Puffer | Im zuge es co2 transports und bildung von bkarbonat entstehen protonen, damit der PH wert nicht abfällt bindet Hb die protonen. ph wert isoelektr. Punkt: isoelektrischen Punkt bei pH 6,8, Jedes Protein hat einen bestimmten pH-Wert, bei dem sich die positiven und negativen Ladungen des Proteins genau ausgleichen |
| Hb Abbau: | findet in der milz statt am mononukleären phagozytensytem, aber auch in der leber Trennung von globin und häm: globin wird zu A.S. abgebaut Entsorgung des Häms ist komplexer: Hämoxygenase ringe des häms werden aufgespalten, Biliverdin, Endprodukt ist Bilirubin Bilirubin wird über blutweg in die leber geschafft, es hängt an albumin=indirektes Im hepatozyt wirds mit aktiver glukuronsäure im gER konjugiert= direktes Das abfallende eisen, wird an die oberfl der makrophagen transportiert, transferrin nimmt sie auf und und transportiert sie zum knochenmark-> werden wieder in Hb eingebaut. |
| Inaktive Hb formen: | CarboxyHb= kohlenstoffmonoxid bindet an Hb mit 300fach höherer affinität als bei sauerstoff, kohlenmonoxid verdrängt aus diesem grund den sauerstoff aus seiner bindung mit Hb Schwindel-> bewusstlosigkeit-> kreislaufzsmbruch->Tod erfolgt durch inneres ersticken Methämoglobin: Hb wird oxidiert, dabei entsteht 3wertiges Eisen Ion. Methb kann genauso wie carboxyhb keinen sauerstoff transportieren |
| Hämoglobinopathien Mutation von Hb. | Das bei Erwachsenen normale tetramere Hämoglobin besteht aus zwei alpha-Ketten und zwei beta-Ketten (alfa beta globulin) Mutiertes β-Globin ist für die Erkrankung der roten Blutkörperchen in Form von Sichelzellenanämie verantwortlich. sichelzellhb hat im reduziertem o2freien zustand eine geringere löslichkeit als normales hb es kommt intrazellulär zur hb ausfällung, dei ery nehmen dabei eine sichelförmige struktur an, folge ist verschluss von gefäßen und eine anämie, weil die erys verstärkt abgebaut werden! |
| Sichelzellanämie wie das Zytoskelett dadurch verändert? | kolloidosmot. Druck im inneren des erys ist höher als der des blutplasmas. Diese druckdifferenz wird durch auswärtstransport von elektrolyten ausgeglichen. blockade der membranpumpen wasser strömt in ery rein, osmotische resistenz der kugelförmigen erys ist verringert verkürzte lebenszeit. |
| Thalassämie: ursache? | Das Fehlen von beta-Ketten verursacht Thalassämie, es werden andere poypeptidketten eingebaut wie z.b. gamma globin-> fetales hb, da hb jedoch in geringen mengen gebildet wird kommts zur mikrozytären hypochromen anämie, kleine erys die wenig hb enthalten! |
| Anämie, Definition | absinken der Hb konz unter 14g/dl beim mann und unter 12 g/dl bei einer frau (Anämien lassen sich nach dem Ery volumen und Hb gehalt im ery einteilen) |
| MCH, MCV, MCHC?? Formeln und werte | MCH= mittleres korpuskuläres Hb= hbmasse innem ery (30pikogramm ist normal) hbkonz/ ery-zahl MCV: mittleres korpuskuläres volumen = eryvolumen (90 femtoliter ist normal) hämatokrit% mal 10/ eryzahl MCHC:mittleres korkpuskuläre hb konzentration (30g/100ml erys) MCH/MCV |
| MCV niedrig, MCH niedrig Was für ne anämie? ursache | Eisenmangel |
| MCV hoch, MCH hoch Was für ne anämie? ursache | Vit B12 Mangel/folsäure mangel |
| Anamieformen | normochrome an- MCH normal – akuter blutverlust hypochrome an- MCH unter 27pg – eisenmangelanämie hyperchrome an- MCH über 34pg- Vit B12 mangel, Folsäuremangel mikrozytäre an- MCV unter 80 femtoliter- eisenmangel, thalassämie makrozytäre an- MCV über 96 fl- vit b12 mangel, folsäuremangel |
| Lipide, allg funktion Klassen? | Energieträger unseres Körpers, Energiereserve für Notzeiten-> sie ersetzen dann die glu als brennstoff stehen unsem organismus genügend andere energieträger wie gluk, a.s. zur verfügung, werden kaum lipide abgebaut gemeinsamkeit aller lipide ist, dass sie aus Acetyl CoA aufgebaut und lipophil sind, lipide lassen sich in 5 klassen einteilen FETTSÄUREN; TAG; PHOSPHOLIPIDE; GLYKOLIPIDE; ISOPRENOIDE |
| Fettsäuren, eigenschaften | sind carbonsäuren, die nen langen schwanz aus kohlenwasserstoffen besitzten. Sie haben sowohl einen hydrophilen (säuregruppe) als auch einen lipophilen(kohlenwasserstffrest) anteil und sind daher amphiphil (fett und wasserlsl.) |
| TAG: eigenschaften, aufbau | TAG: bestehen aus einem glyceringerüst, on dessen 3oh gruppen über eine esterbindung jeweils eine fettsäure gebunden ist, TAGs sind lipophil |
| Phospholipide: | Phospholipide: hauptbstdteil der zellmembranen, gerüst aus glycerin oder sphingosin, einen schwanzteil aus fettsäuren und einen kopfteil aus phosphat mit ner hydrophilen gruppe =amphiphil |
| Glykolipide: | Glykolipide: auch zahlreich in zellmembranen zu finden,haben alle SPHINGOSIN als grundgerüst, fettsäure als lipophilen schwanz, und zucker als hydrophilen anteil, glykolipide sind auch amphiphil z.b. Cerebrosid |
| Isoprenoide | Isoprenoid: leiten sich von isopren ab: terpene-> moleküle mit mehreren isoprenen in ketten hintereinander, dazu zählen vit A,K,E und Ubichinon cholesterin-> entsteht durch faltung einer isoprenkette, aus cholesterin können steroidhormone, calcitrol und gallensäuren hergestellt werden. |
| was können unsere zellen mit lipiden anfangen? | im zentrum des lipidstoffwechsels steht ACETYL COA! Sammelpunkt für abbau und aufbauvorgänge sämtlicher lipide. |
| lipide für den energiestoffwechsel, funktion | für die energiegewinnung sind vorallem fettsäuren wichtig-> können auf und abgebaut werden aber auch in form von TAGs in fettgwb gespeichert werden. diese fettdepots werden bei bedarf abgebaut und die fettsäuren unter großem energiegewinn OXIDIERT! >>beta oxidation, cz, atmungskette oder in leber zu ketonk. Umgewandelt. |
| beta oxidation:funktion | beta oxidation: in mitochondrien, abbau v. fettsäuren zu acetyl coa einheiten, acetyl coa kann in cz eingeschleust werden an den die akette anschließt =ATPHERSTELLUNG |
| Fettsäurebiosynthese: findet wo statt? und funktion?? | im zytosol, ermöglicht den zellen wichtige fettsäuren selbst herzustellen um sie z.b. in membranen einzubauen, überschüssige glukose kann so in fett (TAG) umgewandelt und gespeichert werden (vorallem in leber&fettgwb) aus 8 acetyl-CoAs wird palmitinsäure hergestellt, die umgebaut werden kann zu fettsäuren |
| Biosynthese der TAGs: wozu? | Biosynthese der TAGs: dient der speicherung der fettsäuren im fettgwb, jwls 3 fetsäuren werden mit nem glycerin verbunden und dann als TAG gespeichert |
| Abbau von TAGs: | die lipolyse erfolgt immer dann, wenn der körper vermehrt auf energie angewiesen ist! Lipolyse im Fettgwb: bei nahrungskarenz, TAGs werden durch Triacylglycerinlipase zu fetts. und glycerin gespalten und ans blut abgegeben. Im blut werden die fettsäuren an albumin gebunden -> zur leber ! in der leber werden die fettsäuren zu Acetylcoa einheiten verstoffwechselt(beta ox.) glycerin dient als substrat der gluconeogenese! |
| Lipolyse steht unter hormoneller kontrolle... | bei hohem cAMP spiegel (durch Glukagon,adrenalin,cortisol) wird lipolyse stimuliert steigerung des cAMP spiegels führt nämlich zur aktivierung der proteinkinase A!! diese phosphoryliert die TAG-lipase, welche in phosphoryliertem zustand aktiv ist! |
| Ketonkörper: wann entstehen sie? woraus? wo entstehen sie? warum werden sie gebildet? | in notzeiten wird viel lipolyse betrieben, dadurch gelangen massenhaft fettsäuren in leber, aus deren abbau entsteht sehr viel acetylCOA! Die leber will den energiebedarf der restl. Organe decken, aber acetyl coa kann membranen nicht durchdringen, ketonkörper schon= transportform von acetylcoa Sodass leber anfängt aus acetylcoa k.körper zu bilden! In den organen werden sie wieder in acetylcoa umgewandelt, in den CZ eingeschleust. >>Alle organe(bis auf die leber selbst) können k.körper zur energiegewinnung nutzten! Deshalb sind k.körper lebenswichtiger glukoseersatzstoff |
| Cholesterinfunktion | Phospho und Glykolipide sind die wichtigsten bestandteile unserer zellmembran, manche glykolipide dienen als rez., und manche phosphol. Als intraz. Botenstoffe Cholesterin =membranbaustein, aus ch. Können sämtliche steroidhormone hergestellt werden, calcitrol/vit D wird auch aus ch. Hergestellt, gallensäure ist die ausscheidungsform von ch. Und ist emulgator für die fettverdauung wichtige rolle des ch. Bei entstehung von arteriosklerose |
| LIPIDABBAU | Dummerweise sind fettabbauende enzyme nicht in der lage im lipophilen milieu zu arbeiten, da sie als proteine wasserlösl. Sind! weil die lipide lipophil sind müssen sie emulgiert werden, dadurch bilden sich grenzschichten zw den lipophilen und hydrophilen teilen. Hier können die enzyme die lipide zerlegen, es entstehn lipidbruchstücke, diese bilden mit gallensäuren mizellen. Und werden dann von den enterozyten resorbiert und ans lymphsystem abgegebn! Umgehung des pfortaderkreislauf, direkt transport ins fettgwb, wegen der fkt der lipide als energiespeicher. |
| essezielle lipide | essezielle lipide müssen mit der nahrung aufgenommen werden, da sie nicht selbst syntetisiert werden: die fettsäuren Linolsäure und Linolensäure und die vitamine A,E,K |
| Transport der lipide im blut/ blut verfügt über 2 lipidtransporter: | 1) lipide gebunden an albumin 2) TAGs und Cholesterinester werden von LIPOPRPOTEINEN zu zielorganen transportiert (die hülle bilden amphiphile moleküle wie phosphol o. cholesterin-> sind nach außen hydrophil |
| Hormonelle Regulation des Lipidstoffwechsels: | Glukagon und adrenalin sind für anhebung des blutglu spiegels zuständig, veranlassen leber zur glukoneogenese>> um vermehrt glukoneogenese zu betreiben brauch die leber für sich selbst energie aus der beta oxidation von fettsäuren! also glukagon und adr. Veranlassen auch das fettgwb zur LIPOLYSE insulin sorgt dafür dass lipide im fettgwb gespeichert werden-> hemmt lipolyse, -induziert die extrazellulär lokalisierte Lipoproteinlipase, durch dieses enzym nehmen fettzellen vermehrt fettsäuren &glycerin auf aus denen die TAG synthetisiert werden -induziert fettsäuresynthase |
| Fettsäureabbau | beta oxid. Findet Nur unter aeroben bedingungen In mito statt, in alln zellen außer erys und gehirnzellen, leber und herz beziehen mehr als 50%ihrer energie aus fsäuren aktivierung der fettsäuren im zytosol (mit coenzym a)>> acyl-CoA, danach müssen die fsäuren in mito>> aktiverte fsäuren sind nicht in der lage innere mitochondrienmembran zu durchdringen! Transporter = Carnitin! fettsäurerest wird auf carnitin übertragen= acyl carnitin-> wird im austausch gegen ein freies carnitin molekül in die mitomembran geschleust in der mitomatrix angekommen, wird fettsäurerest wieder auf coenzym a übertragen-> acyl coA betaocidation: abbau von aktivierten fettsäuren zu ACETYLCOA EINHEITEN 1.) oxidation 2) hydratisierung 3.oxidation 4. Thiolytische spaltung Bilanz: 1 molekül acetyl coa, ein nadh+h+, ein fadh2 >>keine direkte atp bildung! |
| ketogenese | ketonl= verbindungen die eine ketogruppe(aceton) erhalten, aber auch betahydroxybutyrat gehört zu ketokörpern ketogenese: nur in mitochondrien der leber, ketok. Werden dann gebildet, wenn acetylcoa nicht mehr komplett durch CZ verstoffwechselt werden kann. 1) 2 moleküle acetyl-coa reagiern zu aceto-acetyl coa 2)acetoacetyl-coa+ acetyl coa= HMG CoA 3) beta-HMG-CoA Lyase-> acetoacetet entsteht 4) !!!!ACETOACETAT wird entweder zu beta-hydroxybutyrat reduziert oder zu Aceton decarboxyliert >> aceton kann aber nicht weiter verstoffwechselt werden! Wird ausgeatmet) bei diabetes mellitus kommts durch gesteigerter ketogenese zu viel aceton abatmung beta-hydroxybutyrat wird an blut abgegeben und dient als energiequelle |
| ursachen gesteigerter kkörperbildung: | längere nahrungskarenz-> blutzucker fällt-> insulin-> lipolyse wird angekurbelt-> viel acetylcoa!-> ketogenese->ketoazidose (ketok. Sind schwache säuren und dissozieern im blut) |
| Ketokörper transport im blut? | Ketonk. Sind im gegensatz zu fettsäuren WASSERLÖSLICH! Und können daher ohne hilfsmittel ins blut , ketoazid: (ketok. Sind schwache säuren und dissozieern im blut) |
| Verwertung der ketok: | können nur von extrahepat. Geweben verwertet werden dabei wird beta hydroxybutyratzu 2 molekülen acetylcoa abgebaut -> diese werden zwecks energieproduktion in CZ/aKette eingeführt |
| Auslöser für Fettsäurebiosynthese im zytoplasma | Nahrungsüberschuss!> organismus baut aus den aufgenommenen kohlenhydraten über acetylcoa neue fettsäurenauf, die er dann als TAG im fettgwb speichert |
| Wie wird aus Pyruvat Acetyl Coa? Warum diese Reaktion?? | Pyruvat gelangt in Mito >>>>Synthese von Acetyl Coa aus Glucose: im mitochondrium! Aka. Pyruvat wird zu Acetylcoa oxidiert durch pyruvatdehydrogense acetyl coa muss vom mito ins zytosol, deshalb reagierts mit oxalacetat zu citrat, citrat kann membran passiern, im zytosol wirds wieder gespalten durch citrat lyase (oxalacetat wird zu malat umgewandelt und geht wieder ins mito) für fettbiosynthese! |
| ACETYLCOA CARBOXYLASE?? | = schrittmacher der fetts.biosynth acetylcoa-> carboxylierung (donator= biotin)zu malonyl coA Carboxylase wird durch aktivierte fetts. Gehemmt, durch citrat, ATP, insulin aktiviert, carboxylase in dephosphorylierter form aktiv Malonyl wird zu Acetyl decarboxyliert Anfallende acylreste werden auf SH gruppen übertragen -> thioesterbindungen kohlenstoffkette wird verlängert für die fettbiosynth. Wird NADPH+H benötigt |
| fettsäuresyntase ablauf | fettsäuresyntase besteht aus 2 identischen UE mit 2 wichtigen SH-gruppen an beide gruppen werden die substrate über Thioesterbindungen gebunden: zentrale SH gruppe- dient zur aufnahme der substrate und bearbeitung der fettsäurekette periphere SH gruppe ablagestelle für die wachsende fettsäurekette |
| TAG synthese: | TAG synthese: die frisch synthetisierten fettsäuren werden in form von TAGs gespeichert Für dessen synthese müssen aber sowohl fettsäuren als auch glycerin aktiviert werden. Fettsäuren werden ATP abhängige reaktion(thiokinase)mit coenzym A zu AcylCoa umgeformt und aktiviert! -aktivierung des glycerins zu glycerin-3-ph (in leber über glycerokinase) Synthese von TaG beginnt mit Acyl Coa und Glycerin-3-pho Gly-3-pho wird mit 2 molekülen acyl coa(unter coa abspaltung)zu phosphatidylsäure verknüpft. phosphatrest wird abgespalten drittes acylcoa wird mit diacylgly verknüpft, fertig |
| Cholesterinsynthese | im zytosol aller kernhaltiger zellen, sehr energieaufwendiger prozess hoher ATP verbrauch als substrat dient acetyl coa, 18 AcetylCoA-> 6 Mevalonsäure C6->6 aktive isopren C5-> 1sqalen C30-> 1lanosterin C30-> 1cholesterin C27 Farnesylpyrophosphat ist ein zwischenprodukt der chol.biosynthese |
| Statine? | Statine zur behandlung eines hohen chol.spiegels hemmen auch die beta HMG-CoA Reduktase und senken somit die cholesterinsynthese |
| Was sind Lipoproteine? Nenne mir lipoproteine | Lipoproteine sind ein transportsystem für lipide im blut. Sie bilden dabei komplexe aus: apolaren lipiden(TAGs Cholesterin) amphiphilen Lipiden(phospholipiden) Proteinen(Apolipoproteinen) bei struktur der lipoproteine unterscheidet man kern und hülle kern: enthält apolare lipide hülle: enthält amphiphilen Lipiden & Apolipoproteine, durch diese bestandteile der hülle wird transport im hydrophilen blut mgl. Chylomikronen, VLDL, IDL, LDL, HDL |
| stoffwechsel der lipoproteine: | Lipide aus der nahrung> über darm resorbiert> in enterozyten zu TAG resyntetisiert> zu chylomikronen verpackt> an lymphe abgegeben>blutkreislauf: im blut nehmen die ChMi von HDL apolipoproteine (ApolipoproteinC2)auf nach fetthaltigen mahlzeiten führen lipopro. Zu einer trübung des blutplasmas, sie schwimmen in den kapillaren der gwb(muskell fettgwb) an die sie sich anheften. an der außenseite der kapillarendothelzellen sitzt lipoproteinlipase!! > durch insulin induziert diese spaltet die TAGs der chylomikronen zu Gly und fettsäuren!! Fetts> werden von extrahepat. Gwb aufgenommen Gly> wird zur leber transportiert> verstoffwechselt ChyMi schrumpfen durch abgabe ihrer lipide zu Ramnants> remnants schwimmen zur leber> weden dort durch rez. Vermittelte endozytose aufgenommen |
| VLDL | VLDL wird in der leber synth., selber abbauweg, nur aus VLDL bildet sich IDL ein teil IDL gelangt zurück zur leber restlicher IDL wird im Blut zu LDL umgewandelt (durch abspaltung aller Apolipoproteine außer Apo B100) |
| LDL | LDL: sehr cholesterinreich, Versorgt periphere gwb mit cholesterin, sehr böse! Wird durch rez.vermittelte endozytose in zielzellen aufgenommen! als ligand für den membrangebundenen LDL-rezeptor dient sein Apo b100 endozytose vesikel mit clathrinmantel |
| HDL | HDL: werden in Leber und Darm synthetisiert, enthalten als lipide vorallem phosphatidylcholin, HDL dienen dem transport von cholesterin aus peripheren gwb, zur leber>> da nur in der leber die cholesterinausscheidung aka. Cholesterinmetabolisierung zu gallensäuren mgl ist |
| wie gelangen HDL zur leber? | Im plasma binden und aktiviern sie das enzym: Lecithin-Cholesterin-Acyltransferase, dadurch wird das cholesterin acyliertCholesterin+ phosphatidylcholin-> cholestinester+ lyso-phosphatidylcholin(diffundiert ab) also gehalt der hdl an cholesterin nimmt zu! Cholesterinreiches HDL gelangt zur leber arterioskleose: hoherLDL spiegel ein risikofaktor (abgabe von cholesterin an blutgefäße), hoher HDL spiegel soll protektiv wirken> sammelt cholsterin ein und transportierts zur leber LDL=böses HDL =butes cholesterin LDL/HDL quotient sollte unter 3 liegen Apoproteine werden in Darm (B48) und in leber (B100) syntetisiert, dienen auch als signalübermittler z.b. als ligand für lipoproteinrezeptoren und sind daher für stoffwechsel der lipoproteine wichtig!! Dichte der lipop. Nimmt mit anteil der apolipoproteine zu! Apolipoprotein E: binbdet rezeptorspezifisch an leberzellen, wichtig für verstoffwechselung TAGreicher bestandteile der chylomikronen Lipide mit esterbindung= verseifbare lipide, weil ester mit starker base reagiert und dann der ursprüngl. Alk entsteht |
| Fettsäuren allg. woraus bestehen sie? ungesättigte/gesättigte unterschied? | nichtverseifbare lipide! Bestehen aus kohlenwasserstoffkette und cooh(carboxylgruppe). Enthält die kohlenstoffkette doppelbindungen so sprichtman von einer einfach oder mehrfach ungesättigter fettsäure. ohne doppelbindungen-> gesättigte fs. |
| Membranfluidität | Einfluss auf die Membranfluidität Membranfluidität wird durch die zahl der doppelbindungen der fettsäuren und durch den gehalt an cholesterin bestimmt. Doppelbindungen, fluidität steigt, cholesterin-> fluidität sinkt! |
| phospholipide aufmalen, welche arten gibt es? | Lecithin->glycerin als grundgerüst Sphingomyelin->sphingosin als grundgerüst |
| wirkweise lipophiler Hormone | lipophile Hormone wirken durch ändeurng der transkription spezifischer gene also auf die dna im zellkern! ihre Rez. Befinden sich intrazellulär. ablauf: lipophiles hormon trifft nukleär oder zytoplasmatisch auf sein intrazelluläres rezeptorprotein dieses rezeptorprotein = regulierbarer transkriptionsfaktor, nur als dimere aktiv, besitzen jwls eine hormonbindungsdomäne und eine dna bindungsdomäne!!!! nach hormonbindung, durchläuft der hormon-rezeptor-komplex eine konformationsänderung> pärchenbildung der rezeptoren der dimerisierte rez, kann nun mit hilfe einer ZINKFINGERDOMÄNE an der großen furche der DNA binden > veränderung der transkrptionsgeschwindigkeit Induktion oder repression dadurch ändert sich die menge des hergestellten enzyms und dadurch auch die zellfunktion >gene der proteine werden verändert abgelesen dementsprechend liegt die zeit zum wirkungseintritt im bereich von stunden! |
| Wirkungsweise Hydrophiler Hormone: | Binden an membranständigen rezeptoren: -G protein assoz. Rez. -1 helixrez (tyrosinkinasen) –ionenkanäle G-Protein gekoppelte Rezeptoren: übertragen extrazelluläre signale auf intrazelluläre signalkaskaden heteotrimeres protein aus alfa,beta gamma UE Hormon bindet an Rez> Konformationsänderung> Hormon-Rez-komplex bindet Gprotein >Austausch von GDP gegen GTP am Gprotein >aktiviertes Gprotein dissoziiert in die GTP bindende alfa komponente alfa UE kann jetzt membranständige effektorenzyme (wie adenylatcyclase oder phospholipase c) aktiviern > dabei enstehn 2nd mssger (cAMP IP3) GTP der alfa UE wird wieder zu GDP gespalten, G protein wird wieder inaktiv > 3 UE des g proteins legen sich wieder zsm |
| Gs/Gq?? | gs –stimuliert adenylatcylase-> cAMP anstieg, gi-> gegenteil adenylacylase ist integraler bestandteil der zellmembran, cAMP kann proteinkinase A aktiviern, proteinkinase a kann serylreste von proteinen phosphorylieren und dadurch den funkrtionszustand dieser proteine ändern= interkonvertierung! Gq- stimuliert phosphokinase c-> anstieg von IP3 und DAG> IP3 öffnet CA2+ kanäle am ER-> anstieg der intraz. Ca2+ konz. -> Ca2+ aktiviert zsm mit DAG die phosphokin C > phosphorylierung von proteinen an serylresten-> funktionsändeurng |
| Signaltransduktion von Tyrosinkinaserezeptoren erklären | Tyrosinkinasen: liegen in der Plasmamembran als monomere vor> hormon bindet an monomer> monomer sucht sich ein identisches rezeptorprotein als bindungspartner (nicht hormonbeladen)> dabei entsteht ein dimer> tyrosinkinaserez nur als dimer aktiv! das wirkungsprinzip dieser rez ist die autophosphorylierung!! autophosphorylierung von intrazellulären tyrosylresten des rezeptors> durch phophorylierung wird der rez. Aktiviert> proteine mit einer SH2 domäne können binden und so aktiviert werden. diese proteine führen zu – konzentrationsändeurng von 2ndmsgern –verändern transkription –können bestandteile des zytoskeletts sein Durch Assoziation mit einem von mehreren Liganden bildet der Rezeptor ein Homodimer bzw. ein Heterodimer, liganden z.b. EGF oder Tumornekrosefaktor bsp. Insulinrez!! |
| was sind Hormone und welche arten gibts? | Hormone sind signalstoffe, die den stoffwechsel ihres zielorgans beeinflussen Einteilung Aminosäurederivate: keine peptidbindung!! T4, T3, dopamin, adr, noradr, serotonin, histamin Peptidhormone:Liberine, Statine,Hypophysenh, Insulin, Glukagon, Parat, Calcitonin, Angiotensin, Gastrin,Sekretin Steroidhormone: Abkömmlinge des Cholesterins, Nebennierenhormone, Geschlechtshormone Fettsäurederivate/Eikasonide: entstehen aus Arachidonsäure, Prostaglandine, Prostazykline, thromboxane, Leukotriene Lipophile Hormone: T4, T3, alle steroidhormone, Eicasonide |
| Katecholamin sythese: | Katecholamine sind A.S.dericate und werden alle aus Tyrosin oder Phenylalanin gebildet synthetisiert werden sie in sympathischen ganglien und im nebennierenmark (v.a. adrenalin) [ausschüttung der katecholamine, durch freisetzung von Ach aus synapse] Syntheseweg: phenylalanin wird hydrxyliert-> tyrosin-> hydrxyliert->DOPA (dihydrxyphenylalanin)->decarboxyliert>biogenes amin Dopamin = erste wirksame form der katecholamine, ist neurotransmitter dopamin> hydroxylierung >noradrenalin > anheften einer methylgruppe> adrenalin die fertigen katecholamine werden in vesikeln gespeichert und bei sympathikusaktivierung ausgeschüttet DIE WIRKUNG AUF DIE ZELLE HÄNGT NICHT VOM HORMON AB SONDERN VON DER ART DES REZEPTORS DER VOM HORMON AKTIVIERT WIRD! |
| adrenerge rezeptoren | alles g protein gekoppelte rez., man unterscheidet alfa 1,2, beta1,2,3(braunes fettgwb) betaRez: steigern den cAMP spiegel>> wirken mit glukagon auf die leberund das fettgwb, beschleunigen lipolyse, glykogenolyse,gluconeogenese alfa2 rez: senken ihn alfa 1 rez: benutzt IP3 weg |
| Sympathikushormone? | Katecholamine sind die Sympathikushormone: phiologische wirkung: erhöhen leistungsfähigkeit des herzens, bronchodilatation, fördern durchblutung der muskulatur metabolisch: mobilisieren glukose und fettsäure werden zu vanillinmandelsäure abgebaut |
| Was ist das Besondere an Ser, Thr, Tyr? | (OH-Gruppe -> Phosphorylierung) |
| essentielle aminosäuren | L-Isoleucin L-Leucin L-Lysin L-Methionin L-Phenylalanin L-Threonin L-Tryptophan L-Valin |
| Ein Superantigen??? | ist ein Antigen, welches ohne Prozessierung eine direkte Bindung von T-Zell-Rezeptor (CD4-positiver T-Lymphozyten) und MHCII-Molekülen (Antigenpräsentierende Zellen) vermittelt. Bei Superantigenen kommt es unabhängig von der Antigenspezifität der T-Lymphozyten zu einer massenhaften unkoordinierten Aktivierung und Freisetzung von Cytokinen, die zu einer fulminanten Entzündungsreaktion des Organismus führt (z.B. Toxic Shock Syndrome) |
| Parathormon | Parathormon ist ein Hormon, das von den Nebenschilddrüsen (Glandulae parathyreoidae) gebildet wird. Reiz für die Freisetzung von Parathormon ist ein Absinken des Kalziumspiegels (Hypokalzämie). Stimuliert osteoblasten, die wiederum osteoklasten aktivieren, diese mobilisieren calsium aus der knochenmatrix: Osteoblasten werden aktiviert> sezernieren M-CSF (makrophagen aktivierend) und RANKL die eine differenzierung von makrophagen zu osteoklasten bewirken! RANKL= mitglied der tumor necrosis factor, wird von osteoblasten gebildet durch bindung von RANKL an seinen spezif. Rezeptor RANK (auf osteoklasten) kommts zur auslösung einer signalkaskade!! Aktivierung von NF-KB (transkriptionsfaktor) gegenspieler von RANKL ist OPG=osteoprotegerin, von osteoblasten sezerniert, bindet an RANKL und die wirkungen neutralisiert gutes RANKL/OPG verhätnis ist wichtig für gleichgewicht das knochenabund aufbaus |
| Calcitonin | Calcitonin ist ein Peptidhormon, das hauptsächlich in den C-Zellen der Schilddrüse gebildet wird. Calcitonin senkt den Kalziumspiegel im Blut. Wenn Calcitonin freigesetzt wird, wird die Aktivität der Osteoklasten gehemmt, wodurch weniger Kalzium aus der Knochensubstanz freigesetzt wird. Außerdem wird die Ausscheidung von Kalzium und Phosphat über die Niere gesteigert. Therapeutisch wird Calcitonin bei der Osteoporose eingesetzt. Calcitriol ist die wirkungsvollste Form des Vitamin D. Es sorgt für die Resorption von Kalzium aus Darm und Niere sowie für den Einbau von kalziumhaltigem Hydroxylapatit in die Knochenmatrix. Ein Mangel an Calcitriol kann zu Osteoporose führen. Eine erhöhter Calcitriol-Spiegel führt zur Symptomatik der Hyperkalzämie. |
| Wie arbeiten Osteoklasten [Säure+ protease]? | Aktivierte oklasten sezernieren protonen und lysosomale proteasen (saure phosphatase)und resorbieren so die knochensubstanz in form von lakunen osteoblasten füllen durch sytnthese von kllg und proteoglykanen, später lagert sich hydroxyapatit an |
| Restriktionsendonukleasen?? | Restriktionsendonukleasen sind bakterielle enzyme, die die dna sequenzspezifisch spalten (dabei enstehen sticky ends o. blunt ends) restriktionsendonukleasen schützen bakterien vor eingedrungener fremder bakteriophagen DNA . Mit restriktionsendonukleasen kann man dna abschnitte gezielt bearbeiten |
| Plasmide?? | Plasmide:ringförmige, doppelsträngige dna abschnitte in bakterien, können z.b. gene für antibiotikaresistenz besitzen aka.a antibiotika spaltende enzyme und zwischen bakterien ausgetauscht werden. Auch die gentechnik nutzt plasmide als transporter(vektoren) für dna sequenzen so ists z.b. menschliche proteine von bakterien produzieren zu lassen! Plasmid (was ist alles enthalten: promotor, zu vervielfältigende gensaquenz) |
| Retroviren und ihre vermehrung Retrovirenaufbau virale onkogene? | retroviren enthalten 1strängige RNA!! Besitzten reverse transkriptase und enthalten virale onkogene bsp. HIV Viren-> Reverse transkriptase: ist ein enzym, dass die virale RNA in cDNA umschreibt =virusreplikation in der zelle! Hierbei wird die RNA abhängige DNA synthese, dann rna abbau, dann die dna abhängige dnasynthese katalysiert die entstandene c-dna wird dann z.b. ins menschl genom integriert, woraufhin die menschl. zelle virale proteine produziert! Auf diese weise können sich die viren vermehren!! Reverse transkriptase: eine RT-PCR ist eine pcr, die rna als ausgangsmaterial benutzt und cDNA syntetisiert Retroviren wie HIV bestehn aus 2 identischen,diploiden RNA strängen, den viralen proteinen, reversen transkriptase, HIV protease, HIV integrase HIV protease: spaltet die neusynthetisierten polypeptide zu Hüll-, struktur- , enzym- und einigen regulatorischen proteinen des HIV auf HIV integrase: dient zur integration der virus DNA in die wirts DNA virale onkogene= genabschnitte= können an jede stelle des menschl. genoms eingebaut werden! |
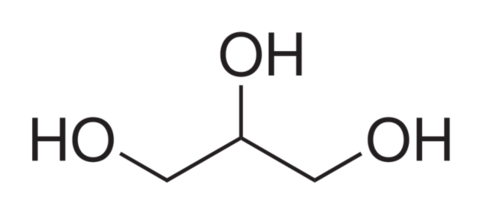{kind=link}
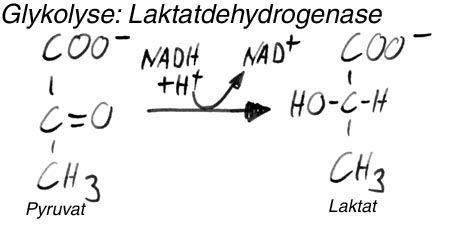{kind=link}
{kind=link}
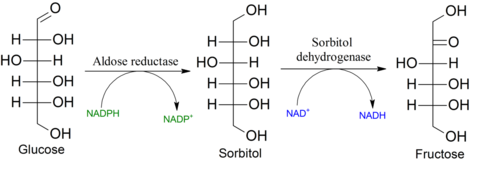{kind=link}
{kind=link}
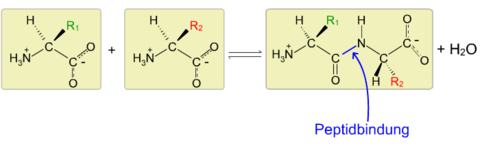{kind=link}
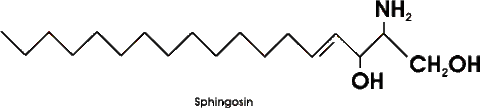{kind=link}
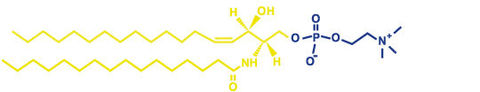{kind=link}
Möchten Sie mit GoConqr kostenlos Ihre eigenen Karteikarten erstellen? Mehr erfahren.