18688814
Descrição
Slides por Fernanda Escalona, atualizado more than 1 year ago
|
|
Criado por Fernanda Escalona
mais de 5 anos atrás
|
|
{kind=link}
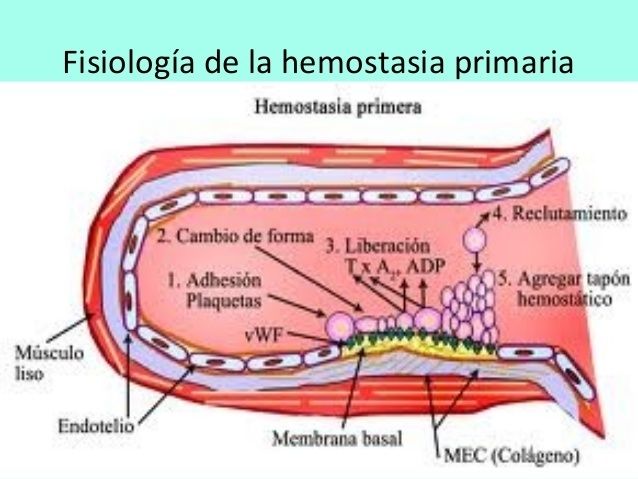{kind=link}
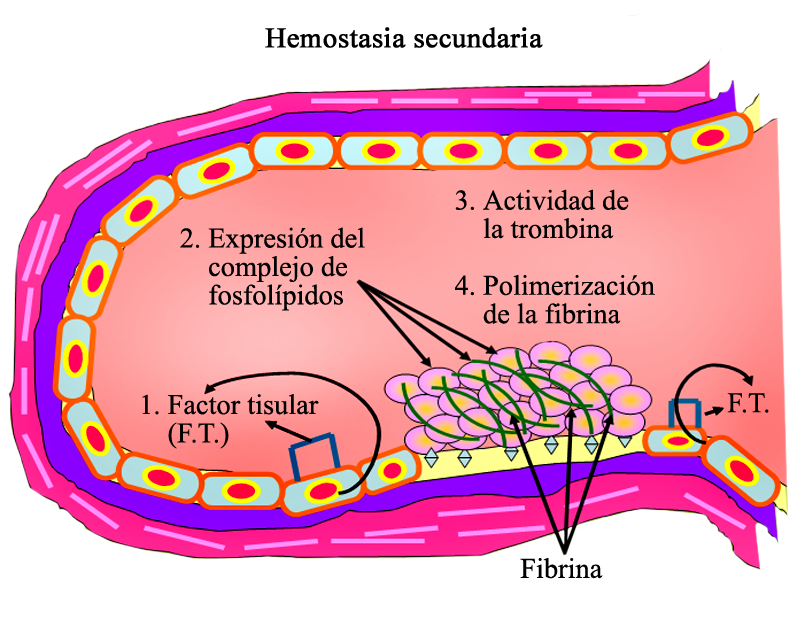{kind=link}
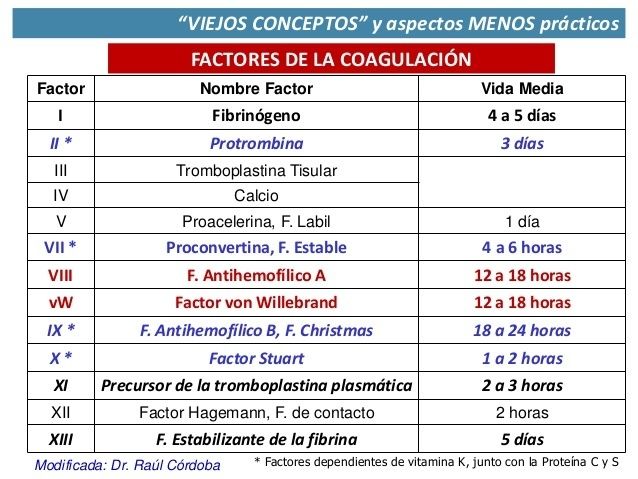{kind=link}
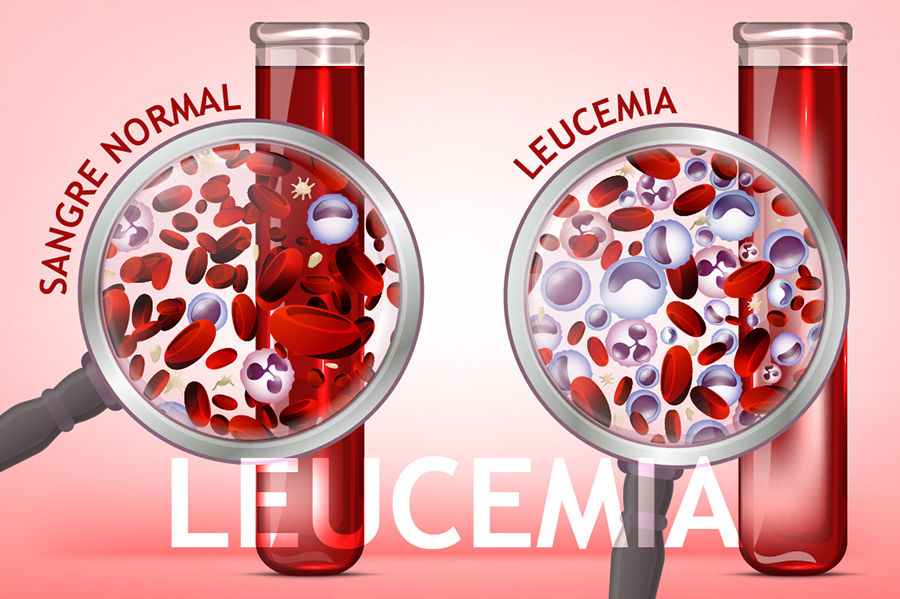{kind=link}
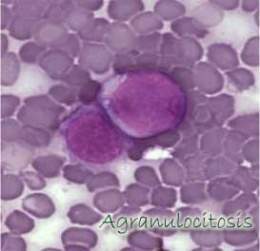{kind=link}
{kind=link}
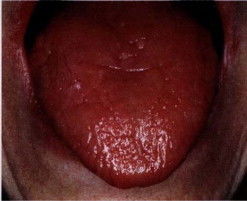{kind=link}
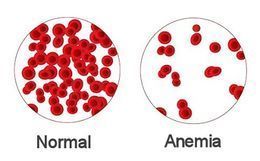{kind=link}
{kind=link}
{kind=link}
Quer criar seus próprios Slides gratuitos com a GoConqr? Saiba mais.