16338160
Beschreibung

Karteikarten von Katja Schmid, aktualisiert more than 1 year ago
|
|
Erstellt von Katja Schmid
vor fast 6 Jahre
|
|
| Frage | Antworten |
| Frage 1: Worin unterscheiden sich Hersteller- und Kundenmarkt? | Unterscheidet sich dadurch wer von beiden Parteien in der besseren Position ist. z.B. Wohnungsmarkt = Hersteller, Kunden = Auto, Fernseher.. |
| Frage 2: Wie lässt sich der Begriff „Qualität“ definieren? | Qualität ist der Überdeckungsgrad aus sowohl expliziten als auch impliziten Anforderungen (“Soll”) einerseits, mit den gelieferten/erreichten Eigenschaften (“Ist”) andererseits |
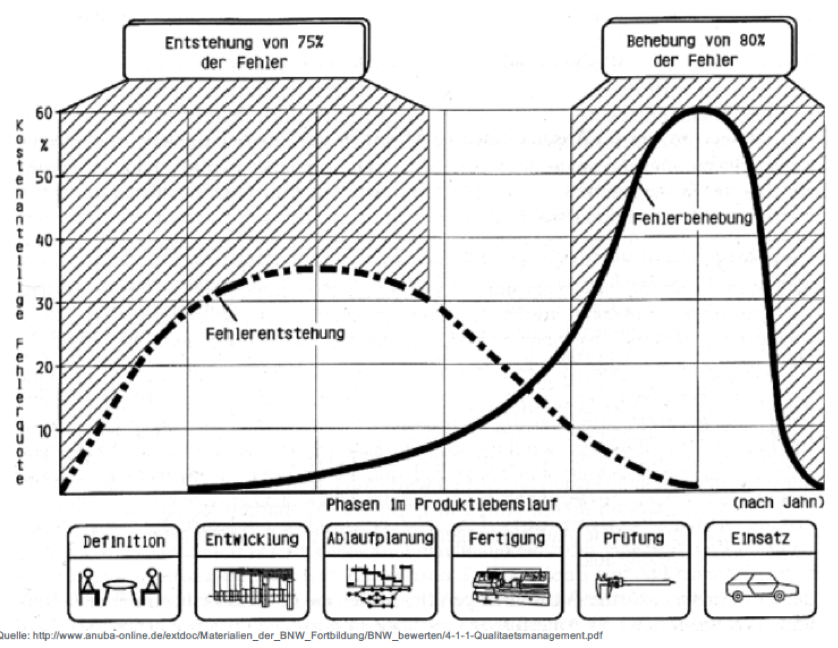
| Frage 3: In welchen Phasen des Produktlebenslaufs entstehen 75% der Fehler? | In den ersten 3 Phasen des Produktlebenszyklus (Definition, Entwicklung, Ablaufplanung) entstehen 75% aller Fehler (hohe kostenanteilige Fehlerquote max 35%). |
| Frage 4: In welchen Phasen des Produktlebenslaufs werden 80% der Fehler behoben? | In den letzten 2 Phasen des Produktlebenslaufs (Prüfung, Einsatz) werden 80% der Fehler erst behoben (hohe kostenanteilige Fehlerquote max 60%). |
| Frage 5: Die ISO 13485 ist in weiten Teilen Deckungsgleich mit einer anderen Qualitätsmanagementnorm. Um welche handelt es sich hierbei? | ISO 9001 |
| Frage 6: Die ISO 9001 fordert Themen wie Kundenzufriedenheit und kontinuierliche Verbesserung des Managementsystems. Durch welche Forderungen und wieso wurden diese in der ISO 13485 ersetzt? | ISO 13485 ersetzt diese Forderungen durch Erfüllung von Kundenanforderungen und der Aufrechterhaltung der Wirksamkeit |
| Frage 7: Normen können im Laufe der Zeit ihre Gültigkeit verlieren. In wessen Schuld liegt es, sich über die geänderten Anforderungen / Rahmenbedingungen zu informieren. | Unternehmen sind selbst verantwortlich, sich über geänderte gesetzliche Rahmenbedingungen zu informieren und diese umzusetzen. Selbiges gilt für Fristen zur Umsetzung von Normen. sogenannte HOL-SCHULD |
| Frage 8: Beschreiben Sie die Begriffe „Verifizieren“ und „Validieren“ in eigenen Worten. | Validierung: Baue ich das richtige Produkt?, Nutzungsziele erreichbar?, können auch User die Zweckbestimmung erreichen? = eine Art Feldexperiment ist, bei dem kontrolliert wird, ob festgelegte Nutzungsziele erfüllt sind und somit die Anforderungen des Kunden auf Tauglichkeit überprüft. Verifizierung: baue ich das Produkt richtig?, Prüfung mit objektiven Mitteln, ob spezifizierte (Produkt-) Eigenschaften erfüllt sind = Bestätigung durch einen objektiven Nachweis, dass Anforderungen erfüllt werden |
| Frage 9: Gelten festgelegte Anforderungen nur an das Produkt selbst, oder auch an die zugehörigen Dienstleistungen? | auch für zugehörige DL |
| Frage 10: Was versteht die ISO 13485 unter einer Kennzeichnung? | jede gedruckte, geschriebene oder graphische Information - auf einem Medizinprodukt oder einem seiner Behältnisse oder sonstigen Verpackungen oder - als Beilage zum Medizinprodukt die sich auf die Identifizierung, technische Beschreibung und Verwendung des Medizinproduktes bezieht |
| Frage 11: Beschreiben Sie den Begriff „Prozess“ in eigenen Worten. | gerichteter, kontrollierter Ablauf von Ereignissen (über gewissen Zeitraum) = system von (zusammenhängenden) Aktivitäten die einen Input in einen Output verwandeln |
| Frage 12: Benennen und beschreiben Sie die einzelnen Phasen des PDCA-Zyklus / Deming-Circle. WICHTIG! | PDCA= Plan, Do, Check, Act Plan: IST-Zustand; Maßnahmen zur Verbesserung eines Prozesses werden geplant; Festlegung welche Ziele erreicht werden sollen und wie diese erreicht werden können, SMART-Kriterien beachten, Prüfkriterien festlegen Do: Beginn der Prozessrealisierung, Prozess entweder auf ganzes Unternehmen entfaltet oder im kleinen Rahmen getestet Check: Ergebnisse der “Do”-Phase werden mit den geplanten Zielen anhand der Prüfkriterien verglichen Act: Auf Basis der “Check”-Phase, Prozess wird reflektiert und dessen Optimierung initiiert, stimmen SOLL und IST überein → Prozess wird standardisiert |
| Frage 13: Wofür steht die Abkürzung SMART bei der Zieldefinition? | Wofür steht die Abkürzung SMART bei der Zieldefinition? S = spezifisch M = Messbar A = anspruchsvoll R = realistisch T = terminiert |
| Frage 14: Was muss bei ausgegliederten Produktionsprozessen, die die Produktkonformität beeinflussen können, beachtet werden? | In der ISO 13485:2016 ist es >nicht mehr ausreichend< , dass die ausgegliederten Prozesse lediglich >erkennbar< sind. Es müssen >jetzt vertragliche Qualitätsvereinbarungen< getroffen werden, die die Lenkung derartiger Prozesse überwacht und sicherstellt. |
| Frage 15: In den USA ist gemäß den Anforderungen der FDA der Device Master Record gefordert. Was ist der Device Master Record und was ist darin enthalten? | = Akte die das Medizinprodukt selbst, aber auch dessen Produktion, Nutzung und Wartung beschreibt - Zweckbestimmung (vgl. 93/42/EWG) - Spezifikationen / Technische Dokumentation (des Geräts und dessen Komponenten) à System-Architektur, Zeichnungen, Berechnungen, etc. - Angaben zur Produktion wie Werkzeuge, Methoden, Verfahren, Spezifikationen / Anforderungen an die Produktion (ISO 13485) - Verfahren zur Qualitätssicherung (z.B. Methoden und/oder Werkzeuge) |
| Frage 16: Was muss bei Nichtanwendung von Anforderungen im Qualitätsmanagement Handbuch beachtet werden? | m QMH können Nichtanwendungen von Prozessen oder Anforderungen begründet werden (z.B. 7.5.9.2 Besondere Anforderungen für implantierbare Medizinprodukte). Hält eine Begründung einer Überprüfung nicht stand, so wird diese als gegenstandslos betrachtet und die Organisation ist zur Umsetzung der Anforderung verpflichtet. |
| Frage 17: Gewisse Dokumente müssen gemäß ISO 13485 gelenkt werden. Ab welchem Zeitpunkt gelten neue und/oder aktualisierte Dokumente für die gesamte Organisation? | Bei der Lenkung von externen Dokumenten, werde diese als Dokumente die für die Planung und den Betrieb des QMS notwendig sind, spezifiziert. In Organisationen können einheitliche Strukturen für Dokumente in Form von Formblättern geschaffen werden ab dem Zeitpunkt wo die neuen Dokumenten allen Mitarbeitern zugänglich sind, z.B. durch Rundschreiben, Aushang usw. |
| Frage 18: Nennen Sie ein Beispiel, wie Sie als Organisation sicherstellen können, dass Änderungen und der aktuelle Überarbeitungsstatus eines gelenkten Dokumentes erkannt werden können. | z.B. Person zur Bewertung von Dokumenten - regulatorisch (MDR, MPG) - normativ (ISO 13485, etc.) Internet, E-Mail, Ankündigung |
| Frage 19: Die ISO 13485 sieht die oberste Leitung in der Verantwortung hinsichtlich der Kundenorientierung. Was muss von der obersten Leitung hierzu sichergestellt werden? | die oberste Leitung muss die Vergabe von Verantwortlichkeiten und Befugnissen sicherstellen (Erfüllung von Kundenanforderungen, Selbstschutzfunktion, Lieferung berücksichtigen) |
| Frage 20: Von wem wird der Qualitätsmanagementbeauftragte ernannt? | von der obersten Leitung |
| Frage 21: Was sind die Aufgaben des Qualitätsmanagementbeauftragten? | - Einführung, Verwirklichung und Aufrechterhaltung der notwendigen Prozesse sicherzustellen - oberste Leitung über die Leistung des QMS und Verbesserungsvorschläge zu informieren - Förderung des QM-Bewusstseins in der gesamten Organisation sicherzustellen |
| Frage 22: Muss die Organisation geeignete Aufzeichnungen hinsichtlich Ausbildung, Schulung, Fertigkeit und Erfahrung für das eingesetzte Personal führen? | Die Anforderungen an personelle Ressourcen werden häufig durch einen „Bewerbungs-Prozess“ sichergestellt und durch “Schulungsprozesse“ weitergeführt. Um sicherzustellen, dass die Personen über die benötigte Kompetenz verfügen, bieten sich Wirksamkeitsnachweise nach einer Schulung an. also nein, es ist nicht explizit notwendig, solche dokumente ergeben sich |
| Frage 23: Welche Verpflichtungen hat die Organisation hinsichtlich der Infrastruktur, die zum Erreichen der Konformität mit den Produktanforderungen erforderlich ist? | Die Organisation muss die Anforderungen an die benötigte Infrastruktur definieren |
| Frage 24: Was fasst die ISO 13485 alles unter dem Begriff „Infrastruktur“ zusammen? | Infrastrukturnachweise sind z.B.: - Gebäude- / Raumpläne - Ablaufdiagramme (Material & Produkt) - Prozesslandkarten - Inventarlisten - IT-Architektur |
| Frage 25: Erfordert die ISO 13485 das Planen und Entwickeln von Prozessen zur Produktrealisierung? | Ja :-) |
| Frage 26: Über welchen „Zeitrahmen“ muss sich das Risikomanagement für Medizinprodukte erstrecken? | Anfang bis absolute Entsorgung, gesamter Produktlebenszyklus Ende= der Moment wo med. Produkt nicht mehr genutzt werden kann Lebensdauer z.B. 8 Jahre lang Kunde versorgen, Instandhaltung etc. -> Sicherheit nur bis dahin garantiert, danach Hersteller nicht mehr Haftbar -> meist mind. 7 Jahre, oft 8 Jahre) |
| Frage 27: Zu welchem Zeitpunkt müssen die Anforderungen im Bezug auf das Produkt bewertet werden? | Die Bewertung muss vor dem Eingehen einer Lieferverpflichtung gegenüber dem Kunden (z.B. Angabe von Angeboten, Annahme von Verträgen oder Aufträgen, Annahme von Vertrags- und Auftragsänderungen) vorgenommen werden. |
| Frage 28: Was muss die Bewertung im Bezug auf die Anforderungen an das Produkt enthalten und wieso hat diese Bewertung für Organisationen auch eine „Selbstschutz“-Funktion? | Inhalt = regulatorischen Anforderungen an Sicherheit und Leistung Ergebnis = freigegebene Zweckbestimmung, Produktbeschreibung oder ein freigegebenes Lastenheft „Eigenschutz“-Funktion = vermieden, dass die Organisation Anforderungen annimmt, welche Sie nicht erfüllen kann / darf. Grund für Eigenschutz = dass, Dinge die nicht in Lastenheft dokumentiert sind, nicht geliefert werden müssen und Herstelle so nicht mehr haftbar ist |
| Frage 29: Welche Entwicklungsmodelle für die Produktrealisierung sind Ihnen bekannt? | - Spiralmodell nach Böhm - V-Modell (Software) - Wasserfall-Modell (FDA) |
| Frage 30: Beschreiben Sie den Begriff „Design Input“ in eigenen Worten. | Design Input bedeutet: „Wir wissen genau, was zu entwickeln ist, wie wir das Produkt realisieren können und wann das Produkt für die Vermarktung bereitstehen wird.“ d.h. wenn Produkt genau durchgeplant ist, nur Aussehen noch nicht |
| Frage 31: Welche Ergebnisse müssen vorliegen, wenn Sie in der Produktrealisierung den Schritt „Design Input“ abschließen möchten? | - vollständiger Projektplan inkl. Budgetschätzung (z.B. angemessene Toleranz: +/- 10%) - Intended Use – eindeutig, prüfbare Produktanforderungen, Akzeptanzkriterien (auch im Bezug auf Normen, Gesetze, Organisation - Realisierungskonzept (ggf. unter Begutachtung externer Experten) - Prozessbeschreibung für Änderungsmanagement (bezogen auf Anforderungen, Ergebnissen etc.) |
| Frage 32: Wieso kann es sinnvoll sein, für die Designvalidierung „Use Case Szenarien“ oder „Benutzerprofile“ zu erstellen? | Um Anwendungsgebiete zu bestimmen, um Sicherheitsmaßnahmen zu erkennen, Produkt-Ausführungen zu definieren, bzw. alle sachen bedenken, die bei jeweiligen Nutzern probleme verursachen würden |
| Frage 33: Was muss eine Organisation festlegen, wenn sie beabsichtigt, im Rahmen einer Beschaffung Verifizierungstätigkeiten beim Lieferanten durchzuführen? | - Verifizierungsplan - Verifizierungsspezifikation - Traceability Matrix - Verifizierungsergebnisse - Zusammenfassung der Ergebnisse in einem Bericht |
| Frage 34: Was sollte bei der Lieferantenauswahl trotz möglicher Zertifizierungen (z.B. nach ISO 13485) beachtet werden? | Lieferanten müssen überwacht und (wieder)bewertet werden neu auch sowie ergreifenden Maßnahmen festlegen, wenn Beschaffungskriterien nicht erfüllt werden Norm ist außerdem kein Garant für erstklassige Qualität. Es sollte also überprüft werden, ob ein Hersteller auch in der Lage ist, die gesetzten Anforderungen umzusetzen. |
| Frage 35: Was versteht man unter einem Los bzw. einer Charge? | Charge = Serie von Waren mit gleichen Eigenschaften, die während eines Arbeitsabschnittes und mit den gleichen Rohstoffen hergestellt, verpackt und mit einer Nummer gekennzeichnet werden bzw. Ladung Los = Gesamtheit der Einheiten verstanden, die in einem Chargenprozess oder einer Losfertigung produziert werden; unter gleichen Bedingungen erzeugt, hergestellt oder verpackt; bzw. „definierte Menge von Stoffen oder eine Anzahl von Medizinprodukten, einschließlich Endprodukt und Zubehör, der/die in einem Verfahren oder in einer Reihe von zusammenhängenden Verfahren verarbeitet wird“ |
| Frage 36: Unter welchen Umständen muss eine Organisation dokumentierte Anforderungen an die Sauberkeit von Produkten einführen? | XX |
| Frage 37: Nennen Sie ein Beispiel, wieso die Platzierung der Produktidentifizierung eine Rolle spielen kann. | Wenn man die Verpackung wegschmeißt, würde man die Produktidentifizierung nicht mehr sehen. → Produkt nicht zuordenbar |
| Frage 38: Wie weit müssen (aktive) implantierbare Medizinprodukte rückverfolgbar sein? | Beauftragte oder Vertriebsmitarbeiter müssen im Hinblick auf die Rückverfolgbarkeit Aufzeichnungen über die Auslieferung von Medizinprodukten führen + ihre Einsicht sicherstellen. Diese Aufzeichnungen müssen Name und Anschrift des Empfängers der Versandverpackung enthalten |
| Frage 39: Wie kann sichergestellt werden, dass nur jene Produkte in Betrieb genommen werden, welche die geforderten Prüfungen zur Freigabe durchlaufen haben? | XX |
| Frage 40: Was kann unternommen werden, damit Produkte mit begrenzter Haltbarkeit oder Produkte die besondere Lagerbedingungen erfordern ihre Konformität mit den Kundenanforderungen auch während der Lagerung / Lieferung aufrecht erhalten? | XX |
| Frage 41: Wie hat die Organisation zu handeln, wenn sie feststellt, dass Messmittel ihre Anforderungen nicht (mehr) erfüllen (scherl)? | Identifikation der betroffenen Produkte. Neuüberprüfung dieser Produkte. Messmittel austauschen/reparieren |
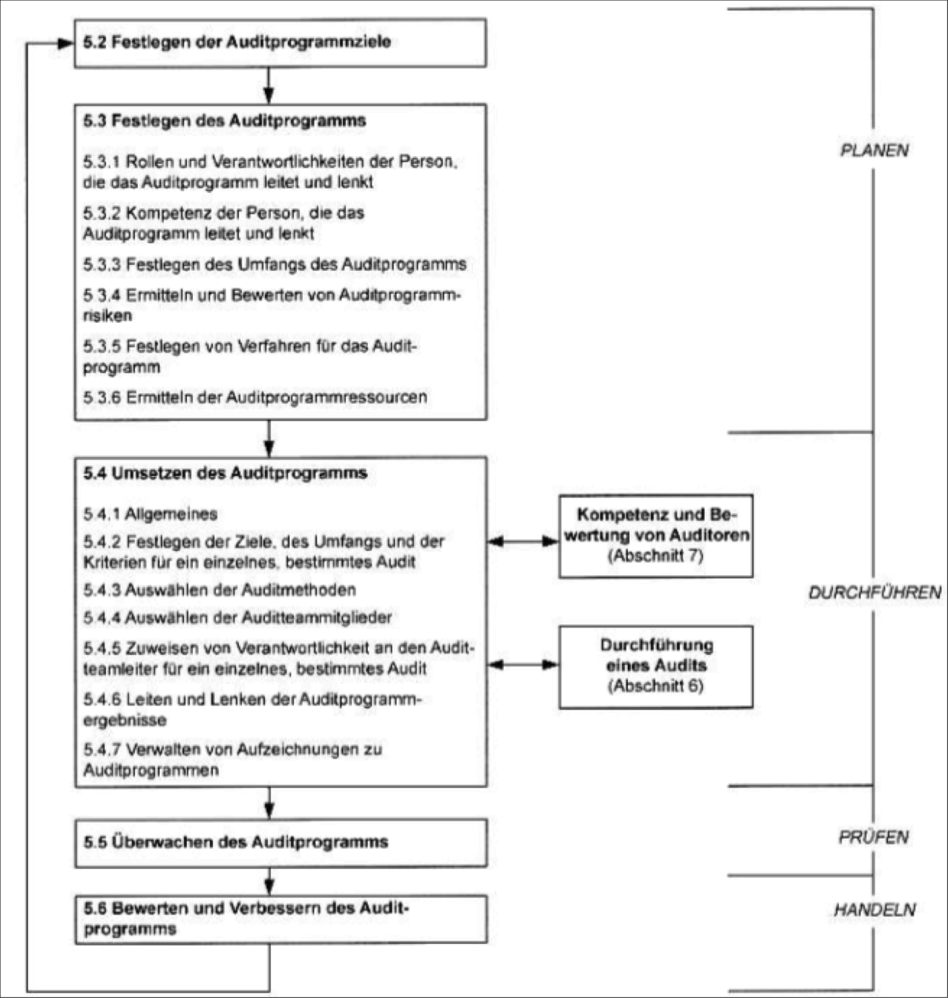
| Frage 42: Die ISO 13485 fordert das Abhalten interner Audits. Welche Parameter sollen bei diesen Audits geprüft werden? | |
| Frage 43: Welche Punkte müssen bei der Auswahl eines Auditors sichergestellt werden? | Um die Objektivität und die Unparteilichkeit zu wahren, dürfen Auditoren ihre eigene Tätigkeit nicht auditieren. |
| Frage 44: Welche Einschränkungen haben Auditoren hinsichtlich ihrer Prüfaufgabe? (Scherl) | sie dürfen nicht ihre eigenen Tätigkeiten(Prozesse) auditieren/prüfen (wenn es dokumentiert wird) |
| Frage 45: Welche Maßnahmen kann eine Organisation ergreifen, um die Inbetriebnahme fehlerhafter Produkte zu verhindern? | Produkte unbenutzbar (gebrauchsunfähig) machen, entsorgen, schreddern bzw. Sperrlager |
| Frage 46: Müssen nachgearbeitete Produkte, den Verifizierungsprozess zur Darlegung der Konformität mit den Produktanforderungen durchlaufen? | Nach Änderungen am Produkt müssen alle Verifizierungsmaßnahmen wiederholt werden, da wechselseitige Abhängigkeiten Einfluss auf die Gesamtspezifikation genommen haben könnte |
| Frage 47: Anhand welcher Kriterien kann sichergestellt werden, dass das QMS seine Eignung und Wirksamkeit aufrecht erhält? | durch eine Managementbewertung, die abhängig von Größe und Struktur der Organisation ca. 1 mal pro Jahr durchgeführt wird es muss nun auch ein verfahren für diese Bewertung aufgestellt und dokumentiert werden |
| Frage 48: Nennen Sie die nationalen Meldefristen, die von Medizinproduktherstellern in Österreich eingehalten werden müssen, sobald sie einen Zusammenhang zwischen einem Produkt und einem Vorkommnis herstellen konnten. | 2 Tage: bei gravierender Gefährdung der öffentlichen Gesundheit 10 Tage: bei Tod oder unerwarteter schwerwiegender Beeinträchtigung der Gesundheit 30 Tage:alle anderen meldepflichtigen Ereignisse |
| Frage 49: Ist die alleinige Fehlerbehebung gemäß ISO 13485 für fehlerhafte Produkte ausreichend? Begründen Sie ihre Antwort. | nein, es muss folgendes getan werden: - Beschreibung des Problems (Änderungsantrag) • Entscheidung (ja / nein / Sofortmaßnahme) - Ursache identifizieren (z.B. Fehler / Nichtkonformität) - Planung der Maßnahmen - Prüfung, Verifizierung, Validierung muss zusätzlich geprüft werden, ob diese Änderung gegebenenfalls auch Einfluss auf bereits fertig entwickelte Medizinprodukte hat |
| Frage 50: Muss die Organisation bereits vor Auftreten eines Fehlers präventiv versuchen, einen Fehler aufzuspüren um diesen vor dessen Auftreten vermeiden zu können? | Ja |
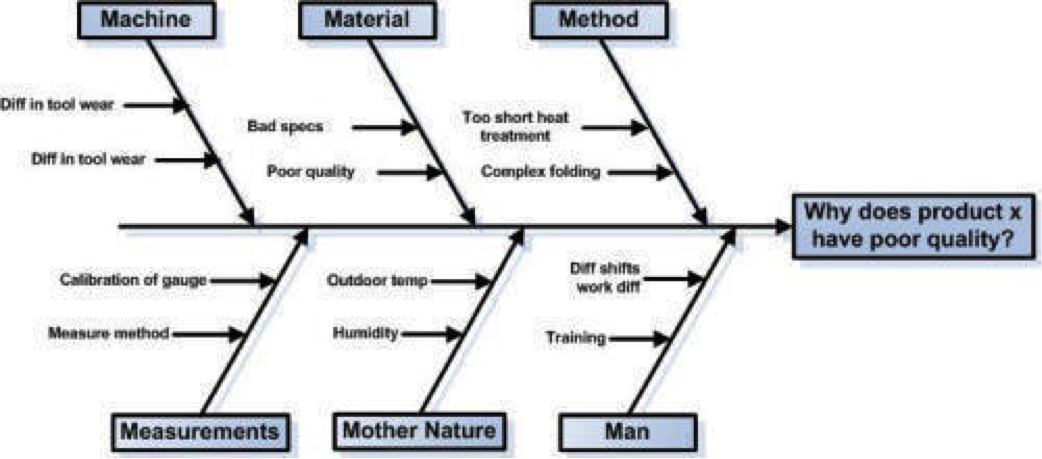
| Frage 51: Nennen Sie die 7 Qualitätsmanagement-Werkzeuge. | Fehlersammelkarte Histogramm Prozessregelkarte Pareto Diagramm Flussdiagramm Korrelations- / Streudiagramm Ishikawa Diagramm |
| Frage 52: Fertigen Sie ein Ishikawa Diagramm basierend auf der 6M-Checkliste und nennen Sie mögliche Einflussfaktoren auf die 6M-Elemente. | |
| Frage 53: Was versteht man unter Poka Yoke? | Ein Prinzip, welches in der traditionellen Vorgehensweise versucht technische Vorkehrungen bzw. Einrichtungen zur sofortigen Fehleraufdeckung und -verhinderung umfasst. Ausgangsbasis: kein Mensch ist in der Lage unbeabsichtigte Fehler zu vermeiden. |
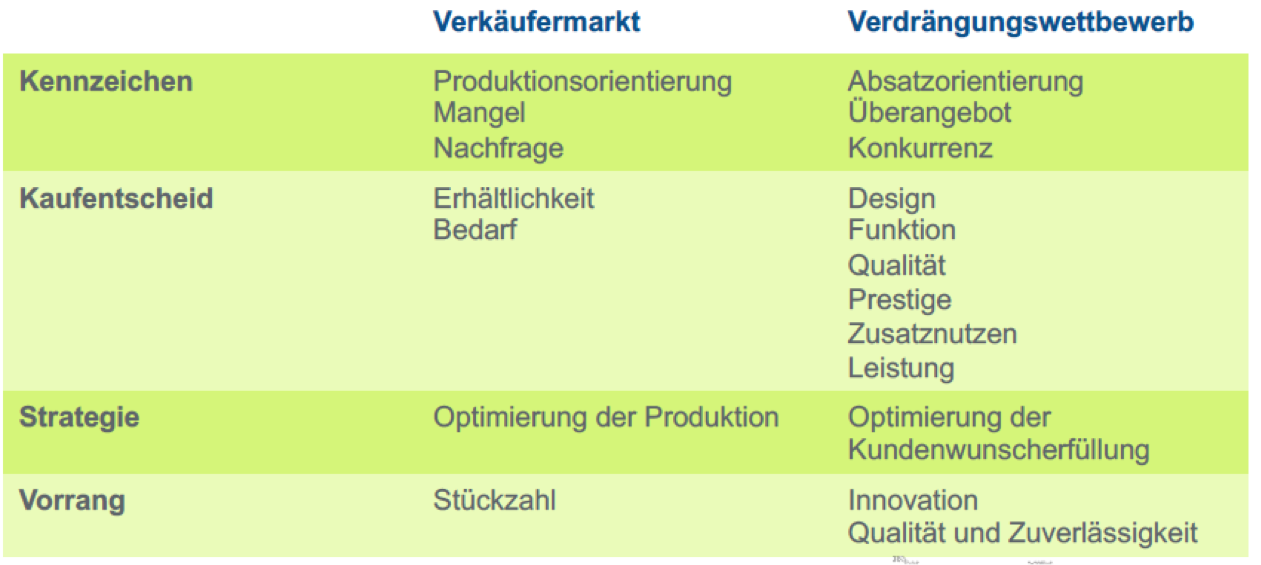
| Frage 54: Wie hat sich der Verkäufermarkt von den 1950er Jahren bis heute verändert? | 1950: Verkäufermarkt (Nachfrage > Angebot) heute: immer mehr Wettbewerbsmarkt bzw. Verdrängungsmarkt (Angebot >>> Nachfrage) |
| Frage 55: Wodurch sind die einzelnen Wettbewerbsphasen seit den 1950er Jahren gekennzeichnet? | Verkäufermarkt: Nachfrage>Angebot Käufermarkt: Angebot>Nachfrage Wettbewerbsmarkt: Angebot>>Nachfrage Verdrängungswettbewerb: Angebot>>>Nachfrage |
| Frage 56: Was sind die Kennzeichen einer Innovation? | … sichern mittel- / langfristig den Unternehmenserfolg … ermöglichen Wettbewerbsvorteil / Gewinnpotentiale … haben Neuheitseffekt … befriedigen die Kundenbedürfnisse … erleichtern die Anwendbarkeit echte Innovation (bietet Lösung die vorher nicht möglich war) unechte Innovation (Differenzierungsversuch zum Wettbewerb, geringer Neuheitswert) |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Möchten Sie mit GoConqr kostenlos Ihre eigenen Karteikarten erstellen? Mehr erfahren.